


Abstract
In humans, 75% of all drugs are metabolized by the cytochrome P450-dependent monooxygenase system. Enzymes encoded by the CYP2C, CYP2D, and CYP3A gene clusters account for ∼80% of this activity. There are profound species differences in the multiplicity of cytochrome P450 enzymes, and the use of mouse models to predict pathways of drug metabolism is further complicated by overlapping substrate specificity between enzymes from different gene families. To establish the role of the hepatic and extrahepatic P450 system in drug and foreign chemical disposition, drug efficacy, and toxicity, we created a unique mouse model in which 30 cytochrome P450 genes from the Cyp2c, Cyp2d, and Cyp3a gene clusters have been deleted. Remarkably, despite a wide range of putative important endogenous functions, Cyp2c/2d/3a KO mice were viable and fertile, demonstrating that these genes have evolved primarily as detoxification enzymes. Although there was no overt phenotype, detailed examination showed Cyp2c/2d/3a KO mice had a smaller body size (15%) and larger livers (20%). Changes in hepatic morphology and a decreased blood glucose (30%) were also noted. A five-drug cocktail of cytochrome P450 isozyme probe substrates were used to evaluate changes in drug pharmacokinetics; marked changes were observed in either the pharmacokinetics or metabolites formed from Cyp2c, Cyp2d, and Cyp3a substrates, whereas the metabolism of the Cyp1a substrate caffeine was unchanged. Thus, Cyp2c/2d/3a KO mice provide a powerful model to study the in vivo role of the P450 system in drug metabolism and efficacy, as well as in chemical toxicity.
Introduction
The mammalian cytochrome P450 system plays a major role in drug metabolism, being involved in the disposition of 75% of all drugs (Schuetz, 2001; Williams et al., 2002; Guengerich, 2008; Tamasi et al., 2011). In humans, 57 P450 genes have been identified (Nelson et al., 2004), five of which, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, are involved in ∼95% of all CYP-mediated drug metabolism (Williams et al., 2004). In contrast, more than 100 putatively functional Cyp genes have been described in the mouse (Nelson et al., 2004). Although the genomic organization of, for example, CYP1A genes is conserved between mice and humans, the CYP2C, CYP2D, and CYP3A gene clusters have diverged significantly between the two species, and assignment of orthologous genes between human and mouse proteins is therefore not possible. The mouse Cyp2c, Cyp2d, and Cyp3a gene clusters contain 15, 9, and 8 functional genes, respectively, compared with only 4, 1, and 4 genes in the corresponding gene clusters in humans (Nelson et al., 2004).
Single Cyp2c, Cyp2d, and Cyp3a knockout mice (Cyp2c KO, Cyp2d KO, and Cyp3a KO) have been used to estimate the in vivo role of Cyp2c, Cyp2d, and Cyp3a enzymes in drug disposition (van Herwaarden et al., 2007; Hasegawa et al., 2011; Scheer et al., 2012a,b; Kazuki et al., 2013). By using this approach, the involvement of mouse enzymes contained within their respective gene clusters has been shown to be responsible for the metabolism of tolbutamide (Cyp2c) (Scheer et al., 2012a); debrisoquine, bufuralol, metoprolol, dextromethorphan (all Cyp2d) (Scheer et al., 2012b); docetaxel, triazolam, and lopinavir) (Cyp3a) (van Herwaarden et al., 2007; van Waterschoot et al., 2009a, 2010). However, surprisingly liver microsomes from Cyp3a KO mice exhibited substantial metabolism of the Cyp3a substrate midazolam and no major difference in drug pharmacokinetics (van Waterschoot et al., 2008). This was explained by the finding that Cyp2c enzymes could also metabolize midazolam, and their expression was significantly increased in the Cyp3a KO animals (van Waterschoot et al., 2008). To create a mouse model in which the role of the cytochrome P450 system in general in drug bioavailability, efficacy, and toxicity in all tissues could be established, we have carried out the deletion of the Cyp2c, Cyp2d, and Cyp3a gene clusters in a knockout mouse line (Cyp2c/2d/3a KO), involving the loss of 30 murine P450 genes. The mice were viable and fertile, although certain phenotypic alterations were observed. The viability of these mice demonstrated that the hepatic P450 isozymes do not have a housekeeping function, and the fact that the mouse line was viable and bred normally will provide a powerful tool for evaluating the role of cytochrome P450s in foreign compound and drug metabolism.
Materials and Methods
Chemicals.
All reagents unless stated were purchased from Sigma-Aldrich (Poole, UK). NADPH was obtained from Melford Laboratories (Ipswich, UK). 7-Benzyloxy-4-trifluoromethylcoumarin (BFC), 7-hydroxy-4-trifluoromethylcoumarin (HFC), 7-benzyloxy quinoline (BQ), and hydroxy-tolbutamide were purchased from BD Gentest (Cowley, UK). Midazolam was obtained from Ninewells Hospital Pharmacy (Dundee, UK). 1′-Hydroxy midazolam, 4-hydroxy midazolam, 1′-hydroxy bufuralol, bufuralol, bupropion, and hydroxy-bupropion were purchased from Toronto Research Chemicals (Toronto, Canada).
Mice.
Cyp2c/2d/3a KO mice were generated from Cyp2c KO, Cyp2d KO, and Cyp3a KO mice described previously (Hasegawa et al., 2011; Scheer et al., 2012a,b) by breeding. Heterozygous offspring from crosses of the single Cyp knockout mice were further bred to generate homozygous Cyp2c/2d/3a KO mice. Cyp2c KO, Cyp2d KO, Cyp3a KO, and Cyp2c/2d/3a KO mice were housed as described previously (Scheer et al., 2008), with ad libitum access to food and water. All animal experiments were carried out under the auspices of the Animal (Scientific Procedures) Act (1986) and after local ethical review at the University of Dundee.
Homozygous 8- to 14-week-old male mice were used for all experiments. Cyp2c/2d/3a KO mice were obtained from TaconicArtemis (Cologne, Germany) and were used for studies at University of Dundee after acclimatization. Cyp3a KO and C57BL/6N WT mice were obtained from Taconic Farms, Inc. (Germantown, NY) and bred at University of Dundee. Mice were killed by a rising concentration of CO2, blood was removed by cardiac puncture into heparinized tubes, and serum was prepared and stored at −20°C until blood chemistry analysis (MRC Harwell, UK). Tissues were removed quickly and either snap-frozen for subsequent preparation of subcellular fractions or fixed in formalin-PBS (4%, GURR, VWR) before transfer to 80% ethanol and processing for wax embedding and sectioning for histopathology.
Preparation of Hepatic and Small Intestinal Microsomal Fractions.
Microsomes were prepared from snap-frozen liver and small intestine tissue harvested from 10-week-old male mice (3 per genotype) as previously described (Finn et al., 2008), and protein concentrations were determined with the BioRad Protein Assay Reagent (BioRad Laboratories Ltd, Herts, UK). NADPH-dependent cytochrome c reduction (Strobel and Dignam, 1978) was used to quantify the activity of P450 oxidoreductase (POR). The P450 content was determined by reduced CO difference spectroscopy (Omura and Sato, 1964).
Immunoblotting.
Immunoblot analysis was carried as previously described (Finn et al., 2008). Fifteen micrograms of liver and 30 µg of small intestinal microsomes were loaded per lane. Immunoreactive proteins were detected using polyclonal goat anti-rabbit or anti-mouse horseradish peroxidase IgG as secondary antiserum (Dako, Ely, UK), and visualized using Immobilon chemiluminescent horseradish peroxidase substrate (Millipore, Watford, UK) and a FUJIFILM LAS-3000 mini imaging system (Fujifilm UK Ltd., Bedford, UK). Densitometric analysis was performed using Multi Gauge V2.2 software (Fujifilm UK Ltd.).
In Vitro Fluorogenic Assay Incubations.
Assays were performed essentially as previously described (Finn et al., 2008) using 20 µg hepatic microsomes and 30 µg of small intestinal microsomes. Reactions were measured in triplicate for each sample (3 mice per genotype) for 3 minutes at the recommended excitation and emission wavelengths for each probe using a Fluroskan Ascent FL fluorimeter (Thermo Labsystems, Cheshire, UK). Turnover rates were calculated using authentic metabolite standards (HQ for BQ; 7-hydroxy-4-trifluoromethylcoumarin for BFC; and resorufin for benzyloxyresorufin, ethoxyresorufin, methoxyresorufin, and pentoxyresorufin (PR) assays.
In Vitro Probe Drug Incubations.
Midazolam 1′- and 4-hydroxylation and tolbutamide hydroxylation assays were performed in triplicate for three samples per genotype as previously described (Finn et al., 2008). Caffeine N-demethylation, bupropion hydroxylation, and bufuralol 1′-hydroxylation assays were carried out in triplicate (3 samples per genotype) using 20 µg of hepatic microsomes and 600, 400, and 400 µM substrate, respectively. Samples were preincubated for 3 minutes at 37°C, and reactions were initiated with a final concentration of 1 mM NADPH and allowed to proceed for 30 minutes before being stopped by the addition of 1 volume of ice cold acetonitrile and ice incubation for 10 minutes. Samples were centrifuged for 8 minutes at 16,000 g to remove particulate material before analysis by LC-MS/MS. Turnover was calculated based on authentic metabolite standards, as detailed below. P values were calculated using an unpaired t test.
In Vivo Pharmacokinetics.
Cyp2c/2d/3a KO and WT mice (n = 4/5 per genotype) were dosed by oral gavage with a five-drug cocktail containing caffeine, bupropion, tolbutamide, bufuralol, and midazolam at 0.5, 3, 0.5, 1, 0.3 mg/ml in water to give final dose of 5, 30, 5, 10, and 3 mg/kg, respectively. Whole blood (10 µl) was taken from the tail vein at intervals after drug administration (10, 20, 40, 60, 120, 240, 360, and 480 minutes) and transferred into tubes containing heparin (10 µl, 15 IU/ml). Samples were stored at −20°C until processed.
LC-MS/MS Analyses of In Vitro and In Vivo Pharmacokinetic Samples.
Samples were thawed, and 95 µl of acetonitrile containing 0.2 µg/ml of triazolam (internal standard, electrospray ionization positive) plus 0.2 µg/ml of chlorzoxazone (internal standard, electrospray ionization negative) was added. Samples were shaken for 20 minutes before centrifugation at 16 g for 10 minutes. The supernatant was then analyzed by UHPLC/MS-MS using a Waters Acquity Ultra High Performance Liquid Chromatography (Micromass, Manchester, UK) and Micromass Quattro Premier mass spectrometer (Micromass) with Electrospray detection. The chromatography was performed using a Kinetex 1.7µ C19 100A; 50 × 2.1 mm column (Phenomenex, Macclesfield, UK) with mobile phases of 0.1% formic acid in water (A) and acetonitrile and 0.1% formic acid. A linear gradient at a flow rate of 0.5 ml/min was run over 2 minutes as follows: 0–0.25 minutes: 95–67% A; 0.25–1.0 minutes: 67–63% A held for a further 0.5 minute then returning to initial conditions for a final 0.5 minute. The cone voltage and collision energy were optimized for each substrate and multiple reaction monitoring data were acquired (Supplemental Table 1). Concentration of parent drug and metabolite were calculated using authentic standards.
Analyses of In Vivo Pharmacokinetic Data.
Pharmacokinetic parameters were calculated using the WinNonLin software, v3.1. A noncompartmental model was used to calculate area under the curve (AUC), terminal half-life, maximum plasma concentration (Cmax), and clearance (Cl). Data were then used to calculate P values using an unpaired t test.
Sample Preparation for Proteomic Analysis.
Frozen liver tissue was thawed by addition of 10 volumes SDS-DTT-Tris lysis buffer (4% SDS, 0.1 M Dithiothreitol, 100 mM Tris-HCl, pH 7.6) and then homogenized by rotor-stator (2 × 5 seconds at 20k revolutions). Homogenate was heated to 95°C for 5 minutes, sonicated (2 × 5 seconds), and then centrifuged at 16,000 g for 10 minutes. Supernatant (protein sample for analysis) was removed, aliquoted, and stored at −80°C until use. Protein samples, comprised of 10 μg of experimental sample mixed with 10 μg of 13C6-lysine metabolically labeled spike-in reference standard, were diluted in Laemmli sample buffer and electrophoresed a short distance into a 12% bis-tris gel in 4-morpholinepropanesulfonic acid running buffer supplemented with antioxidant (Life Technologies, Paisley, UK). After staining with Coomassie blue, each gel lane was cut into three bands and processed by the in-gel trypsin digest and peptide extraction method of Schevchenko and colleagues (2006). Peptides sample concentration was determined by Nanodrop (Thermo Fisher Scientific, Waltham, MA) and adjusted to 0.2 mg/ml in water containing 0.1% (vol/vol) trifluoroacetic acid.
LC-MS/MS Analysis of Peptide Samples.
A nanoflow liquid chromatograph (Agilent 1200, Agilent, Santa Clara, CA) with a LTQ-Orbitrap XL (Thermo Fisher Scientific) was used to analyze the protein digests. Approximately 0.4 μg of total peptide was loaded onto a trap column at a flow rate of 10 µl/min for 3 minutes, and the flow was then reversed to an Agilent Zorbex nano C18 column (0.0075 mm internal diameter; 15 cm; 3 µm particle size). The peptides were resolved with a 3-hour binary gradient at a flow rate of 300 nl/min as follows: 0% Buffer B for 5 minutes followed by 2–30% Buffer B for 140 minutes, 30–90% Buffer B for 15 minutes, 90–0% Buffer B for 10 minutes, and 0% Buffer B for 10 minutes. Buffer A contained 2% acetonitrile and 0.1% formic acid in water, and Buffer B contained 0.1% formic acid in acetonitrile. The column was periodically cleaned with a 2-µl injection of buffer containing 50% acetonitrile and 0.1% formic acid in water. A Proxeon nanospray source with a stainless steel emitter (Thermo Fisher Scientific) was used to interface the Agilent nanoLC and LTQ-Orbitrap. Spray voltage was set at 1.8 kV. The Orbitrap was tuned using Glu-Fibrinogen B peptide. Full scans between 300 and 2000 atomic mass unit (in Orbitrap) and data-dependent MS/MS with top 6 precursor ions (2+ to 4+ charged) in LTQ were employed. Orbitrap was operated in profile mode at the resolution of 30,000 or 60,000 with a lock mass set at 445.1200 [polycyclodimethylsiloxane (Olsen et al., 2005)], and LTQ was operated in a centroid mode with isolation width = 1 (m/z), normalized collision energy = 0.25, and activation time = 30 ms. A dynamic exclusion of 30 seconds was used to maximize the acquisition of MS2 on peptides with lower intensity.
Proteomic Data Analysis.
Data were processed using MaxQuant [version 1.3.0.5 (Cox and Mann, 2008)] with the International Protein Index mouse database (version 3.68) and the integrated Andromeda search engine. Cysteine carbamidomethylation was used as a fixed modification, with N-terminal acetylation and methionine oxidation as variable modifications. The peptide false discovery rate was set to 0.01%, minimum peptide length was 7, and a maximum of 2 miscleavages was allowed. Only unique peptides were used for quantification, with the requantify option left unchecked. Relative quantification of experimental samples was achieved by reference to the spike-in standard “heavy” sample, with fold changes calculated using the ratio-to-ratio approach (Monetti et al., 2011). For calculation of statistical significance, normalized values were log2-transformed and then analyzed by two-way analysis of variance with Bonferroni correction for multiple comparisons using Prism 6 (Graphpad, La Jolla, CA).
Results
Generation and Phenotypic Analysis of Cyp2c/2d/3a KO Mice.
The development and characterization of single Cyp2c KO, Cyp2d KO, and Cyp3a KO mice has been described previously (Hasegawa et al., 2011; Scheer et al., 2012a,b). These mice lack 14 of 15 Cyp2c genes, all 9 Cyp2d genes, and 7 of 8 Cyp3a genes. Mouse Cyp2c44 and Cyp3a13 genes, both located at some distance from the main gene clusters, were not deleted. Cyp2c/2d/3a KO mice were generated from the single Cyp gene cluster knockout mice by breeding. No impairments in breeding performance or deviations from the expected Mendelian ratio of genotypes were observed during the process of generating the Cyp2c/2d/3a KO line.
The triple gene cluster null mice were viable, fertile, and exhibited no gross anatomic abnormalities. However, at 10 weeks of age the mice were smaller than wild-type (WT) animals (24.2 ± 0.9 versus 28.3 ± 2.0 g; P = 0.0001, n = 8 of each genotype). Post mortem examination of Cyp2c/2d/3a KO mice revealed a slightly paler than normal liver, which had a distinct mottled appearance. No other tissues exhibited any other macroscopic morphologic changes. A small increase in liver weight (13%; from 1.26 to 1.43 g, P = 0.005, n = 8 of each genotype) was observed in Cyp2c/2d/3a KO mice, which translated into a significant increase in liver-to-body weight ratio (30%; 0.045 ± 0.005 versus 0.059 ± 0.003; P ≤ 0.001, n = 8 of each genotype).
Pathologic examination of hematoxylin and eosin (H&E)-stained liver sections from Cyp2c/2d/3a KO mice (Fig. 1) showed a zonation in staining, which would account for the mottled appearance of the liver. This appeared to be due to changes in hepatocytes in zone 1 (portal) versus zones 2/3. Interestingly in zone 1, the hepatocytes gave the appearance of being smaller and more densely packed than normal (arrows), with the result that Kupffer cells (and perhaps also including endothelial cells) in this region appeared to be more numerous than in surrounding tissue (Fig. 1). In contrast, the hepatocytes in zones 2 and 3 gave the appearance of being "expanded" such that the cells were larger and less dense than normal. There was no evidence of steatosis in these sections. H&E-stained liver sections from WT mice looked essentially normal. These phenotypes were not observed in the single cluster knockout mouse lines.
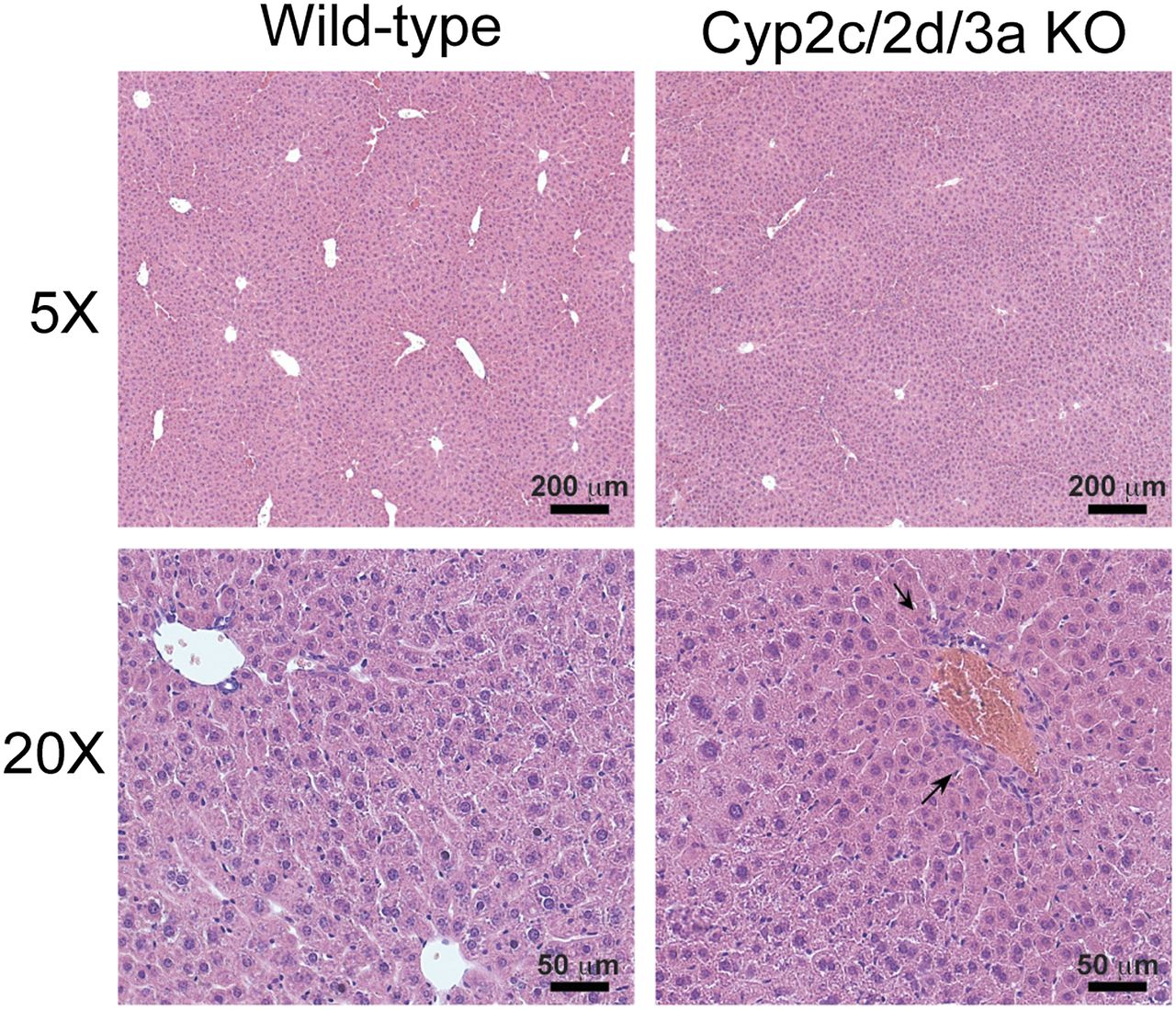
Liver pathology in Cyp2c/2d/3a knockout mice. Representative hematoxylin and eosin stained liver sections from WT and Cyp2c/2d/3a KO mice. Arrows show regions in Cyp2c/2d/3a KO mice where hepatocytes appear smaller and/or more densely packed. Photomicrographs representative of n = 4 or 5; bright field, magnification ×5 and ×20.
Blood Chemistry.
Analysis of blood chemistry of untreated Cyp2c/2d/3a KO and WT mice showed no significant differences between genotypes for alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, creatinine, cholesterol (total, low-density lipoprotein, or high-density lipoprotein), free fatty acids, triglycerides, total bilirubin, or lactate dehydrogenase (Supplemental Fig. 1); however, blood glucose was significantly decreased in Cyp2c/2d/3a KO mice compared with WT controls (13.3 ± 1 versus 19.1 ± 2.5 mmol/l; P = 0.02). The large error bar on the lactate dehydrogenase data were due to one Cyp2c/2d/3a KO mouse having a value approximately fourfold higher than the others, possibly as a result of hemolysis in this sample.
Analysis of Cytochrome P450-Dependent Monooxygenase Components.
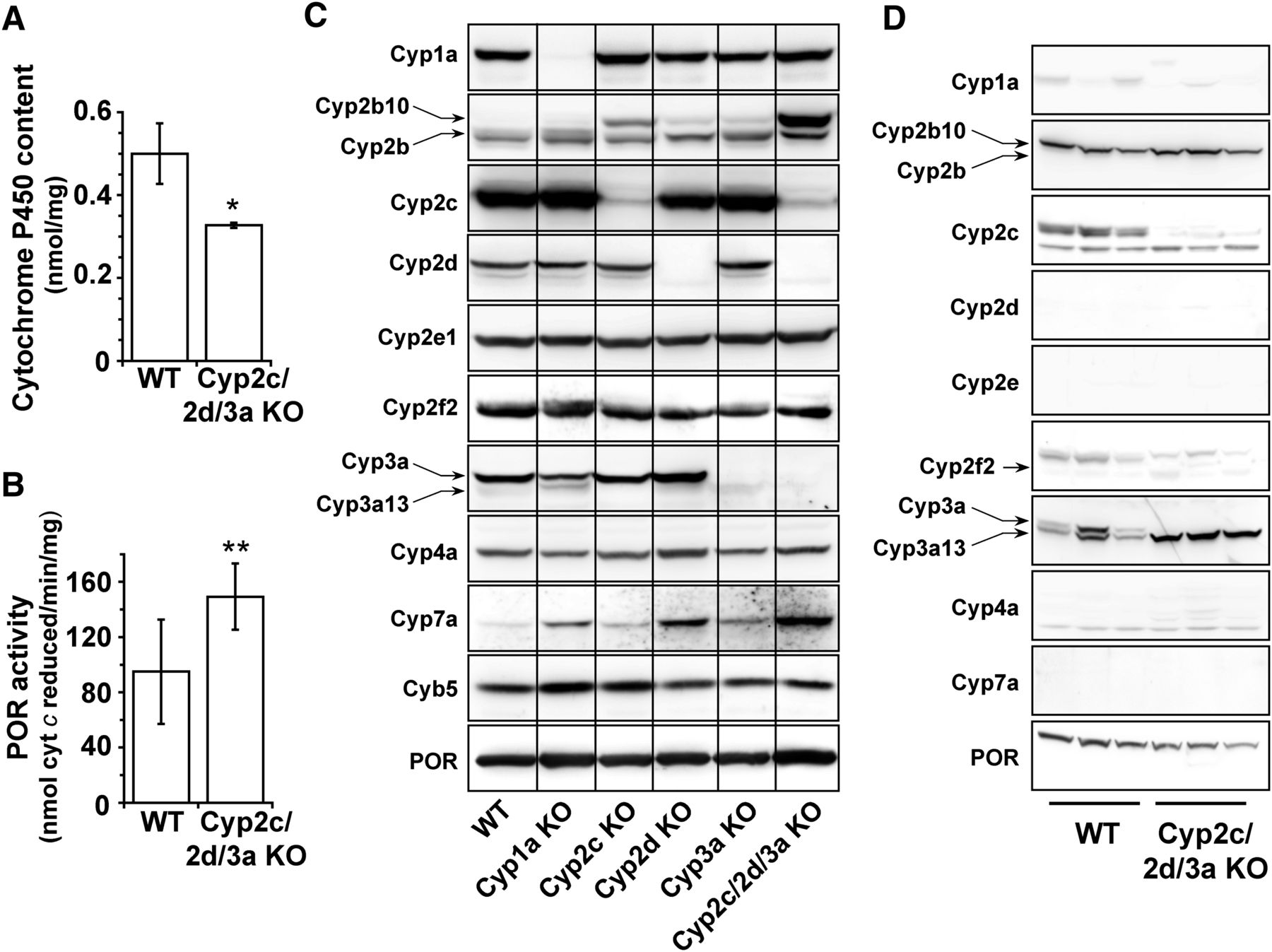
Interestingly, despite having deleted 30 P450 genes, total cytochrome P450 content in the liver was reduced by only 36% (Fig. 2A), whereas POR catalytic activity was significantly increased, albeit less than twofold relative to WT values (Fig. 2B) and consistent with increased POR protein expression on immunoblot (Fig. 2C) and by LC-MS/MS proteomic analysis (Supplemental Fig. 2). Expression of Cyp2c, Cyp2d, and Cyp3a proteins seen in hepatic microsomes from WT mice was not observed in the single or triple Cyp cluster KO animals; this also demonstrates Cyp3a13 and Cyp2c44 are essentially absent from untreated mouse liver. A slight increase in the expression of Cyp2b10 was observed in each of the single Cyp cluster KO mice but was more pronounced in Cyp2c/2d/3a KO mice (20-fold higher than WT) (Fig. 2C). This increase, however, was substantially less than that typically observed after phenobarbital treatment (Ross et al., 2010). Similarly, Cyp7a, involved in cholesterol and bile acid biosynthesis, was increased in all three single Cyp cluster KO mice but to a higher level in the compound KO (Fig. 2C). The proteomic analysis showed very slight increases (<2-fold) for Cyp1a2, Cyp2e1, and Cyb5 (Supplemental Fig. 2), although the latter was not reflected in the immunoblot (Fig. 2C).

Cytochrome P450 isozyme and P450 reductase expression in WT, single Cyp cluster KO, and Cyp2c/2d/3a KO mice. (A) Comparison of hepatic cytochrome P450 levels as measured by CO difference spectroscopy (n = 3 for each genotype). (B) Hepatic cytochrome P450 oxidoreductase activity in samples from WT and Cyp2c/2d/3a KO mice (n = 3 performed in duplicate for each genotype). *P ≤ 0.05, **P ≤ 0.01 (C) Western blotting of hepatic microsomes (15 µg per lane) from WT Cyp1a KO, Cyp2c KO, Cyp2d KO, Cyp3a KO, and Cyp2c/2d/3a KO mice was carried out as described in Materials and Methods (n = pools of 2–4 animals per genotype). (D) Western blotting of small intestinal microsomes from WT and Cyp2c/2d/3a KO mice. Each lane represents an individual mouse (30 µg per lane).
In the small intestine, expression of Cyp1a, Cyp2d, Cyp2e, Cyp2f2, Cyp4a, and Cyp7a proteins were low or undetectable in both WT and Cyp2c/2d/3a KO mice (Fig. 2D). Cyp2b10 and POR were expressed in this tissue, and deletion of the three Cyp gene clusters had no effect on their expression. Cyp2c and Cyp3a (upper band in Cyp3a blot) were undetectable in Cyp2c/2d/3a KO mice; however, interestingly, there was an increase in the levels of a crossreacting protein, probably Cyp3a13. This change was not observed when the Cyp3a gene cluster was deleted alone (Hasegawa et al., 2011).
Measurement of In Vitro Cytochrome P450 Activities Using Fluorogenic Probes.
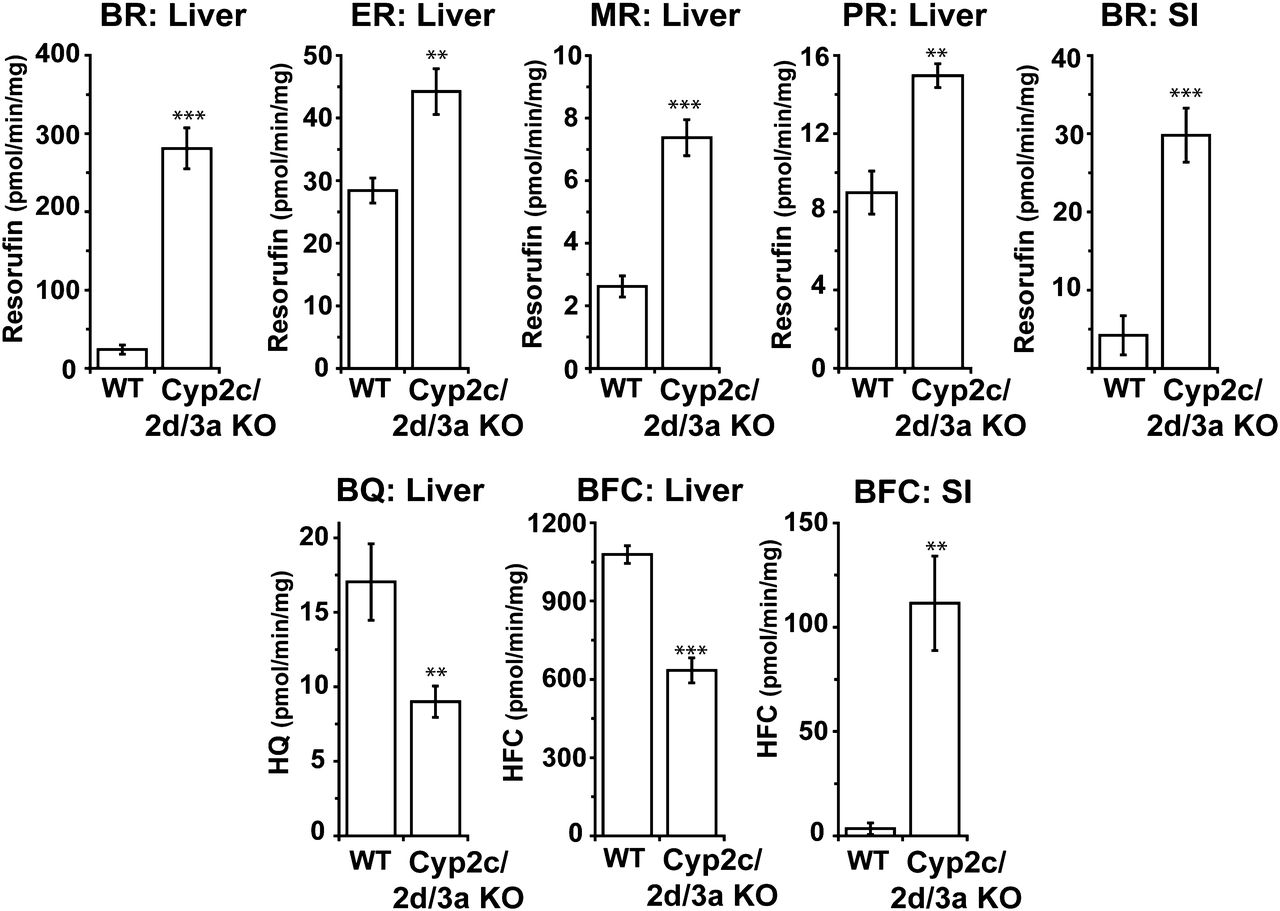
Liver and small intestinal microsomes from WT and Cyp2c/2d/3a KO mice were incubated with fluorescent substrates, which have been described as P450 isozyme probes to establish the effect of deleting the Cyp2c, Cyp2d, and Cyp3a gene clusters on their activity (Fig. 4). Ethoxy-and methoxyresorufin, reported as probes of the Cyp1a subfamily in rats (Burke et al., 1985), showed a 1.5- and 2.8-fold increase in turnover in hepatic microsomes from Cyp2c/2d/3a KO mice; because the activities were still very low and the finding that there was no change in the levels of Cyp1a protein, these increases can be attributed to the increase in either POR or Cyp2b10 expression (Fig. 2, B and C). Small intestinal microsomes did not metabolize either ethoxy- or methoxyresorufin. Benzyloxyresorufin is metabolized by a number of rodent P450 isozymes, including Cyp2b10 in the mouse (Chatuphonprasert et al., 2012); liver samples from Cyp2c/2d/3a KO mice showed an 11.8-fold increase in turnover, which can be accounted for by the increased expression of this protein (Fig. 2C). Small intestinal microsomes from Cyp2c/2d/3a KO mice also showed an increase in the rate of benzyloxyresorufin metabolism. This cannot be attributed to Cyp2b10 (Figs. 2D and 4) but could be explained by the increase in Cyp3a13 in this tissue (Hagemeyer et al., 2010). The Cyp2b substrate pentoxyresorufin (Chatuphonprasert et al., 2012) showed only a small increase in turnover (1.7-fold) in hepatic samples from Cyp2c/2d/3a KO mice compared with WT controls, suggesting that P450s other than Cyp2b10 (Fig. 2C) are responsible for the metabolism of this substrate (Fig. 4).
The isoform specificities for 7-benzyloxyquinoline (BQ) and 7-benzyloxy-4-trifluoromethylcoumarin (BFC) are not well characterized in mice; it has been suggested that Cyp3a11 can metabolize both substrates. There are, however, data from studies in rat and human samples that members of other CYP families can contribute to the metabolism of these compounds (Stresser et al., 2002). The decrease in BQ and BFC metabolism in the Cyp2c/2d/3a KO mice (48 and 42%, respectively) is consistent with loss of the main Cyp3a11-dependent metabolism (along with potential Cyp2c and Cyp2d contributions), while indicating that other murine cytochrome P450s (possibly Cyp1a and Cyp2b) also contribute to BQ and BFC turnover (Fig. 4). BQ was not metabolized in small intestinal microsomes; however, a significant (33-fold) increase in activity was observed with BFC, potentially attributable to the increased Cyp3a13 expression, although overall activity was extremely low (Figs. 2D and 4).
Measurement of In Vitro Cytochrome P450 Activities Using Probe Drugs.
Metabolism of a number of P450 probe drugs by hepatic microsomes from WT and Cyp2c/2d/3a mice is shown in Fig. 3. Metabolism of the Cyp1a probe drug caffeine (Buters et al., 1996; Casley et al., 1997) to the hydroxylated metabolite paraxanthine was significantly increased (3.3-fold) in the Cyp2c/2d/3a KO mice as compared with WT, which was similar to that seen with the fluorogenic probes ethoxy- and methoxyresorufin (Fig. 4). Bupropion hydroxylation, a marker of Cyp2b (Molnari et al., 2011), was increased (40-fold) in the absence of the three Cyp gene clusters, which could be accounted for by the induction of Cyp2b10 protein (Fig. 2C). It should be noted, however, that the metabolism of this substrate in WT mice was very low. Activity toward the Cyp2c probe drug tolbutamide (DeLozier et al., 2004; Finn et al., 2008; Scheer et al., 2012a) was reduced 76% in the Cyp2c/2d/3a KO mice; the residual activity indicates that other murine P450s play a role in its disposition. Bufuralol 1′-hydroxylation, which is attributed to Cyp2d-mediated metabolism (Scheer et al., 2012b), was virtually absent in Cyp2c/2d/3a KO mice, with only 3% of WT activity remaining. The two hydroxylation pathways of the CYP3A4 probe midazolam exhibited were affected differently in the Cyp2c/2d/3a KO samples; 1′-hydroxylation was strongly reduced (66%), whereas the 4-hydroxy metabolite was slightly increased (20%).

Cytochrome P450-mediated in vitro metabolism of probe drugs using hepatic microsomes from WT and Cyp2c/2d/3a KO mice. Assays for metabolism of caffeine, bupropion, tolbutamide, bufuralol, and midazolam were performed with liver microsomes from WT and Cyp2c/Cyp2d/Cyp3a KO mice as described in Materials and Methods. Bars represent mean ± S.D. (n = 4 or 5 animals per group; with each sample run in triplicate). For the midazolam data, the white bars represent the 1′-hydroxy and the black bars represent the 4-hydroxy metabolite. Statistical significance comparing the data from Cyp2c/2d/3a KO samples to that generated from WT samples for each metabolite, *P ≤ 0.05, ***P ≤ 0.001.

Cytochrome P450-mediated in vitro metabolism of probe substrates using hepatic and small intestinal microsomes from WT and Cyp2c/2d/3a KO mice. Assays for metabolism of benzyloxyresorufin (BR), ethoxyresorufin (ER), methoxyresorufin (MR), pentoxyresorufin (PR), 7-benzyloxyquinoline (BQ), and 7-benzyloxy-4-trifluoromethylcoumarin (BFC) were performed with liver and small intestinal (SI) microsomes from WT and Cyp2c/Cyp2d/Cyp3a KO mice as described in Materials and Methods. Bars represent mean ± S.D. (n = 4 or 5 animals per group; with each sample run in triplicate). Statistical significance comparing Cyp2c/2d/3a KO to WT mice, with **P ≤ 0.01, ***P ≤ 0.001.
In Vivo Pharmacokinetics of a Five Drug Cocktail.
WT and Cyp2c/2d/3a KO mice were dosed orally with a five-drug cocktail containing caffeine, bupropion, tolbutamide, bufuralol, and midazolam as probes for Cyp1a, Cyp2b, Cyp2c, Cyp2d, and Cyp3a, respectively, and blood samples collected by serial tail vein bleeding over the course of 8-h postgavage. The pharmacokinetic data are shown in Fig. 5, Table 1 and Supplemental Table 3. For caffeine, although both Cmax and AUC were decreased in Cyp2c/2d/3a KO mice, these did not reach statistical significance. The pharmacokinetics of the paraxanthine metabolite showed the half-life to be the only parameter significantly altered, down 40% in Cyp2c/2d/3a KO mice compared with WT animals. The oral bioavailability of bupropion was significantly decreased in Cyp2c/2d/3a KO mice, consistent with increased expression of Cyp2b10 (Fig. 2C). The Cmax of the parent drug was reduced by 78% compared with WT, whereas clearance and half-life were 8.6-fold faster and 2.4-fold longer, respectively, in Cyp2c/2d/3a KO mice. There was also an 87% reduction in AUC for bupropion; the AUC of the hydroxylated metabolite of this drug was increased (1.7-fold) compared with WT animals.

In vivo pharmacokinetic profiles and parameters of caffeine, bupropion, tolbutamide, bufuralol, and midazolam and the resulting metabolites in WT and Cyp2c/2d/3a KO mice. Drugs were administered orally as a five-drug cocktail as described in Materials and Methods to WT (n = 5), Cyp3a KO (n = 3), and Cyp2c/2d/3a KO (n = 4) mice: ○, WT; □, Cyp3a KO (for midazolam); ●, Cyp2c/2d/3a KO. Data are mean ± S.E.M.
Pharmacokinetic parameters for drug cocktail administered to WT and Cyp2c/2d/3a KO mice
A five-drug cocktail was administered to WT (n = 5), Cyp3a KO (n = 3), and Cyp2c/2d/3a KO (n = 4) mice and blood samples were taken for analysis and drug levels determined as detailed in Materials and Methods. Pharmacokinetic parameters were derived from the WinNonLin software package, and data are shown as mean ± S.D.
The elimination of tolbutamide was significantly reduced in the Cyp2c/2d/3a KO animals. Because the AUC0–8h:AUC0–inf ratio was 0.2, well below the accepted cut-off of 0.8 for accurate prediction of the extrapolated pharmacokinetic parameters, AUC0–inf, Cl, and half-life were not calculated (Gabrielsson and Weiner, 2007) (Table 1). The Cmax for tolbutamide in Cyp2c/2d/3a KO mice was decreased by almost half compared with WT, although there was no change in AUC0-8h between the two genotypes. The AUC0-8h of the hydroxy tolbutamide metabolite was decreased 4.8-fold in Cyp2c/2d/3a KO animals.
Bufuralol was cleared extremely rapidly by WT mice (3.7 l/min/kg), and deletion of the three Cyp gene clusters had a profound effect on its pharmacokinetics. Similarly to tolbutamide, this resulted in an AUC0–8h:AUC0–inf ratio of less than 0.8 (0.7), so only Cmax and AUC0–8h are reported for this drug (Table 1). Cmax was increased 15-fold and AUC0–8h by 26-fold in the Cyp2c/2d/3a KO mice compared with WT. The AUC of 1′-hydroxy bufuralol metabolite was 12.3-fold lower in Cyp2c/2d/3a KO mice.
Midazolam was also included as a probe drug, because in the single Cyp3a KO, its pharmacokinetics are unchanged because of a compensatory change in Cyp2c55 expression (van Waterschoot et al., 2008). In the Cyp2c/2d/3a KO animals, Cmax and AUC were increased 3.3- and 4-fold, respectively, compared with WT mice, and clearance was reduced to 28% of WT rates, whereas no change in half-life was observed. Compared with Cyp3a KO mice, the Cmax and AUC of midazolam were both increased 2.1-fold, and clearance was reduced by 65% in the triple Cyp cluster knockout animals. The plasma concentration of 1′-hydroxy midazolam was reduced in the Cyp2c/2d/3a KO mice, with a 67 and 78% decrease in Cmax and AUC, respectively, compared with WT and a 50 and 31% decrease, respectively, compared with the Cyp3a KO mice. Overall, the levels of 4-hydroxy midazolam produced were one order of magnitude lower than the 1′-hydroxy midazolam metabolite. The pharmacokinetic profiles observed in the WT and Cyp3a KO mice were similar, whereas in the Cyp2c/2d/3a KO the area under the curve was significantly increased (4.3-fold) compared with WT.
Discussion
The cytochrome P450-dependent monooxygenase system has evolved as a multigene family of proteins with the capacity to insert an atom of molecular oxygen into a substrate and to carry out a variety of other reactions. This property has been exploited through evolution to enable the P450 system to play a pivotal role in xenobiotic and drug metabolism, metabolic homeostasis, and a wide range of other functions (Kakizaki et al., 2008; Konno et al., 2008; Schmidt et al., 2011; Wang et al., 2013). Although there is a significant delineation of substrate specificity between cytochrome P450 isozymes involved in xenobiotic metabolism and those involved in endogenous metabolic processes, certain xenobiotic metabolizing enzymes such as those in the CYP2C, CYP2D, and CYP3A gene families have been associated with a number of endogenous functions (Supplemental Table S2). In this paper we describe the generation and characterization of a novel triple Cyp2c, Cyp2d, and Cyp3a gene cluster knockout mouse model with a deletion of 30 functional cytochrome P450 genes. To our knowledge, this is the first report of a transgenic mouse model in which such a large number of genes have been deleted simultaneously. Interestingly, the Cyp2c/2d/3a KO mice were viable and fertile and appeared overtly phenotypically normal, demonstrating that the deleted mouse Cyp2c, Cyp2d, and Cyp3a genes collectively have no essential housekeeping function(s) but rather fulfill adaptive endogenous roles. Some phenotypes were, however, observed. At 10 weeks of age, the mice were smaller and liver weights slightly increased, resulting in a higher liver-to-body weight ratio, and livers from Cyp2c/2d/3a KO mice appeared morphologically different from WT, which was associated with modest irregularities in hepatic zonation. It should be noted that a liver phenotype has also been reported for the single Cyp2c KO model (Scheer et al., 2012a) but not for the Cyp2d KO or Cyp3a KO lines from which the Cyp2c/2d/3a KO mice were derived. Therefore it appears likely that these changes are a consequence of the deletion of the mouse Cyp2c genes. Interestingly, some of the findings described in the Cyp2c KO mice, such as a decreased alkaline phosphatase activity, high-density lipoproteins, and cholesterol were not observed in the Cyp2c/2d/3a KO line; the reason for this is currently unknown and may be a consequence of changes specific to the complex knockout line and warrants additional investigation. In contrast, the small, but significant, decrease in blood glucose in the Cyp2c/2d/3a KO mice has not been reported for any of the underlying single Cyp gene cluster knockouts. The mechanistic rationale underlying these observations is unclear and is an intriguing subject for further study. No other significant changes in blood chemistry parameters or in macroscopic tissue morphology were found.
We have described single knockouts for these gene clusters previously (van Herwaarden et al., 2007; Hasegawa et al., 2011; Scheer et al., 2012a,b; Kazuki et al., 2013), and many of the phenotypes observed in these models are also reflected in the compound gene cluster deletion. However, in the case of the single Cyp3a gene cluster knockout (van Waterschoot et al., 2008, 2009b), the reported compensatory induction of Cyp2c55 could not occur because this gene is now deleted. Although the potential for compensatory change in cytochrome P450 gene expression in the KO model is significantly reduced, it has not been completely eliminated. For example, the intestinal expression of Cyp3a13, which was not deleted in the model, and the hepatic expression of POR and Cyp2b10 in particular were all increased. Comparison of hepatic P450 levels between the Cyp2c/2d/3a KO model and the individual cluster knockouts (Fig. 2C; Supplemental Fig. 2) showed very few differences in P450 expression when analyzed collectively, the exception being that the increases in Cyp2b10 and Cyp7a were more pronounced. Cyp2b10 is essentially not expressed in the liver of WT mice, and the mechanism underlying its increased expression remains to be established. However, in a number of previous gene deletions—the hepatic P450 reductase null (HRN) and cytochrome b5 null (BCN) models (Henderson et al., 2003; McLaughlin et al., 2010)—we also observed an induction of hepatic Cyp2b10 expression. In the HRN model, the altered P450 expression appears to be due to changes in the hepatic ratio of saturated to unsaturated fatty acids as a consequence of hepatic steatosis (Finn et al., 2009); however, steatosis was not observed in the Cyp2c/2d/3a KO mice. This finding also demonstrates that the steatosis is not a consequence of the loss of Cyp2c, 2d, and 3a gene function. Surprisingly, despite the deletion of 30 genes from the three major Cyp gene clusters, total P450 content was only reduced by 36%. The induction of Cyp2b10 may in part explain this; however, in all likelihood, Cyp2b10 only constitutes a small proportion of the residual P450 expression. The absence of clear changes in any other significantly expressed P450s in mouse liver, such as Cyp2f2 (Jenkins et al., 2006), suggest that there are many important functions, other than in drug metabolism, of the hepatic P450 system yet to be discovered.
The Cyp2c/2d/3a KO mouse line provides a powerful tool to study the role of hepatic and extrahepatic P450s in drug metabolism and chemical toxicity. One interesting finding from the use of this model (Fig. 4) is that that the previously reported specificities of the fluorescent substrates for particular P450s are not precise. For example, the dealkylation of pentoxyresorufin, a putative Cyp2b substrate, increases by less than 50% in Cyp2c/2d/3a KO mice despite a significant induction of Cyp2b10, whereas benzyloxyresorufin metabolism, reported to be carried out by a number of P450s including Cyp2b isoforms, is increased by more than threefold (Fig. 2C). Furthermore, benzyloxyresorufin metabolism is increased by a similar amount in small intestinal microsomes from Cyp2c/2d/3a KO mice, despite there being no change in Cyp2b expression (Figs. 2D and 4). BQ and BFC have both been reported to have selectivity for Cyp3a isoforms (at least in human and rat) and are only partially decreased in the liver of the KO mice.
In vivo, the metabolism of the respective Cyp2c, Cyp2d, and Cyp3a probe substrates tolbutamide, bufuralol, and midazolam was markedly affected in the triple knockout (Fig. 5; Table 1). The results obtained for midazolam are of particular interest. Despite midazolam being a Cyp3a substrate and in agreement with previous results (van Waterschoot et al., 2008), we found similar concentrations of the parent compound and its 1′- and 4-hydroxy metabolites in WT and single Cyp3a cluster KO mice. This finding is attributed to a compensatory increase in Cyp2c enzymes (van Waterschoot et al., 2008). This was confirmed by the finding that midazolam concentration was significantly increased and the production of 1′-hydroxy midazolam attenuated in the Cyp2c/2d/3a KO mice carrying both Cyp2c and Cyp3a deletions. The formation of 4-hydroxy midazolam was, however, increased in the Cyp2c/2d/3a KO mice. This is probably because of other murine P450s, for example Cyp2b10, being involved in the generation of this metabolite.
The slightly decreased in vivo metabolism of caffeine in Cyp2c/2d/3a KO mice is not reflected in the rate of production of paraxanthine in vitro, which is increased, suggesting that there are other pathways of disposition involved. Cyp1a2 has been shown to play a major role in caffeine disposition (Buters et al., 1996). The increase in in vitro turnover is likely to be due to increased Cyp1a and POR expression in these samples; however, in an in vivo setting this effect may be diluted by any extrahepatic activity and secondary metabolism of the metabolite. This illustrates the difficulties that may be encountered when trying to directly extrapolate a pharmacokinetic outcome from in vitro data. The increased in vitro metabolism of bupropion by hepatic microsomes from Cyp2c/2d/3a KO mice (Fig. 3) corresponded well with the decreased bioavailability of the drug in vivo and supports the specificity of Cyp2b isoforms for bupropion.
Cyp3a13 and Cyp2c44, which are both located at a significant distance to the main Cyp3a and Cyp2c gene clusters on the respective chromosomes, were not deleted in the Cyp2c/2d/3a KO model. Cyp2c44 could not be detected in our proteomic analysis of the liver and its expression in other tissues is unknown (DeLozier et al., 2004). Cyp3a13 is expressed at very low levels constitutively in the liver (Yanagimoto et al., 1997), and no increase in expression was observed in the KO mice. The apparent increased intestinal expression of Cyp3a13 should be considered when using this model.
In previous work we demonstrated the potential of the HRN and RCN (liver/gut) P450 reductase KO models to establish the role of the P450 system in drug disposition and toxicological response (Pass et al., 2005; Finn et al., 2007; Arlt et al., 2011). The absence of Cyp2c, Cyp2d, and Cyp3a enzymes in all tissues of the compound cluster knockout model, with the result that the mice contain only a limited number of P450 genes involved in xenobiotic metabolism in any tissue, has a wide range of applications in drug development. These include defining the role of the P450 system in drug disposition and in the toxicological effects of development compounds so that the toxic liability may be designed out. The model can also be applied in efficacy studies, by increasing the plasma levels of drugs with poor bioavailability due to metabolism by the P450 system. Furthermore, comparison of pharmacodynamic profiles between WT and Cyp2c/2d/3a KO mice may be used to establish whether a drug and/or metabolite(s) have pharmacological activity, whereas the deletion of P450 function will allow the role of Phase II metabolites—often not observed because of high P450 activity in rodents—to be more clearly defined in drug disposition.
Acknowledgments
The authors thank Anja Graaff, Roman Kaesbach, Oliver Dahlmann, Sarah Neu, Heiko Cziudaj (TaconicArtemis), and Julia Carr (University of Dundee) for technical assistance.
Authorship Contributions
Participated in research design: Scheer, Henderson, and Wolf.
Conducted experiments: McLaughlin, MacLeod, and Henderson.
Contributed new reagents or analytic tools: Scheer, Rode, and Macleod.
Performed data analysis: McLaughlin, MacLeod, and Henderson.
Wrote or contributed to the writing of the manuscript: Scheer, McLaughlin, MacLeod, Henderson, and Wolf.
Footnotes
- Received February 28, 2014.
- Accepted March 26, 2014.
Program support came from Cancer Research UK [Grants C4639/A10822 to R.W.] and from ITI Life Sciences, Scotland.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the concentration-time curve
- BFC
- 7-benzyloxy-4-trifluoromethylcoumarin
- BQ
- 7-benzyloxyquinoline
- Cl
- clearance
- Cmax
- maximum plasma concentration
- ER
- ethoxyresorufin
- HRN
- hepatic reductase null
- KO
- knockout
- LC-MS/MS
- high-performance liquid chromatography-tandem mass spectrometry
- P450
- cytochrome P450
- POR
- cytochrome P450 oxidoreductase
- WT
- wild type
- NADPH
- nicotinamide adenine dinucleotide phosphate (reduced)
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}