


Abstract
The cytochrome P450 (P450) 1 family is an important phase I enzyme involved in carcinogen activation. Nitidine chloride (NC) is a pharmacologically active alkaloid with polyaromatic hydrocarbon found in the roots of Zanthoxylum nitidum (Roxb.) DC, a traditional medicinal herb widely used in China. We examined the inhibitory effects of NC on CYP1A1, 1B1, and 1A2. NC significantly inhibited CYP1A1- and 1B1-catalyzed ethoxyresorufin O-deethylation activity (IC50 = 0.28 ± 0.06 and 0.32 ± 0.02 μM, respectively) in a concentration-dependent manner, but only showed slight inhibition of CYP1A2 activity (IC50 > 50 μM). Kinetic analysis revealed that NC competitively inhibited CYP1B1 with a Ki value of 0.47 ± 0.05 μM, whereas NC caused a mixed type of inhibition on CYP1A1 with Ki and KI values of 0.14 ± 0.04 and 0.19 ± 0.09 μM, respectively. The observed enzyme inhibition neither required NADPH nor revealed time dependency. Molecular docking manifested the generation of strong hydrogen-bonding interactions of Ser116 in CYP1A1 and Ser127 in CYP1B1 with methoxy moiety of NC. Additionally, NC-induced alteration of estradiol (E2) metabolism was also investigated in the present study. Hydroxyestradiols, including 2-hydroxyestradiol [(2-OHE2) nontoxic] and 4-hydroxyestradiol [(4-OHE2) genotoxic] generated in recombinant enzyme incubation systems and cultured MCF-7 cells were analyzed, and NC was found to preferentially inhibit the nontoxic 2-hydroxylation activity of E2 mediated by CYP1A1. In conclusion, NC was a mixed type inhibitor of CYP1A1 and a competitive inhibitor of CYP1B1. The remarkable inhibition on E2 2-hydroxylation might increase the risk of 4-OHE2–induced genotoxicity.
SIGNIFICANCE STATEMENT CYP1 enzymes catalyze oxidative metabolism of a variety of compounds and are known to play a crucial role in the development of cancer. CYP1A1 and CYP1A2 are responsible for hydroxylation of estradiol (E2) at the C-2 position, resulting in the formation of 2-OHE2, which is proposed to be a detoxification pathway. However, CYP1B1-mediated hydroxylation of E2 at the C-4 position has been suggested to be a tumor initiator. The present study found that nitidine chloride is a mixed type inhibitor of CYP1A1 and a competitive inhibitor of CYP1B1. NC preferentially inhibited the nontoxic E2 2-hydroxylation pathway mediated by CYP1A1, which might increase the risk of 4-OHE2–induced genotoxicity and cause severe drug-drug interactions.
Introduction
Zanthoxylum nitidum (Roxb.) DC as a traditional herbal medicine is mainly distributed in Asian nations (Liao et al., 2013; Li et al., 2017). The roots of the plant have been used extensively in China for treatment of various diseases, including neuralgia, toothache, rheumatism, and swelling of the throat (Liu et al., 2017). Meanwhile, it also usually serves as a natural ingredient in commodities such as toothpaste. An abundance of alkaloids has been isolated and identified from herbal roots, and nitidine chloride (NC) (Scheme 1) is the major one of them (Arthur et al., 1959). The reported pharmacological activities of NC include antihuman immunodeficiency virus (Tan et al., 1991), anti-inflammatory (Hu et al., 2006; Wang et al., 2012), antimalarial (Gakunju et al., 1995; Bouquet et al., 2012), and antitumor agent (Iwasaki et al., 2010; Pan et al., 2011; Chen et al., 2012; Fang et al., 2014; Ou et al., 2015).

Chemical structures of NC (A), ERF (B), and E2 (C).
Cytochromes P450s (P450s) are heme-containing enzymes primarily responsible for oxidation or reduction of the majority of pharmaceutical agents currently in use (Foti and Dalvie, 2016). Comprised of a vast number of isoforms, this gene superfamily of proteins mostly belongs to intrahepatic enzymes (Vrba et al., 2004). The CYP1 family plays a crucial role in the phase I metabolism of endogenous and exogenous compounds such as estrogen and polyaromatic hydrocarbon and is known to be involved in the generation of carcinogens (Lo et al., 2013; Lee et al., 2016). Furthermore, CYP1 members exhibit differences in tissue distribution. Constitutive expression and production of CYP1A2 occur in human liver, whereas CYP1A1 and 1B1 are mainly localized in extrahepatic tissues (Lo et al., 2013).
Estradiol (E2) (Scheme 1) is a natural estrogen with many physiologic effects (Lozan et al., 2017). Oxidation of E2 to 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2) mediated by CYP1A1, 1B1, and 1A2 has been suggested to be associated with detoxification and tumorigenesis, respectively (Lakhani et al., 2003; Lee et al., 2003). Many CYP1 inhibitors have been demonstrated to share several structural motifs that are planar with two or more hydrophobic aromatic rings (Zanger and Schwab, 2013; Lee et al., 2016). For example, sanguinarine is identified to be a noncompetitive inhibitor of CYP1A1 and a competitive inhibitor of CYP1A2 (Vrba et al., 2004). Berberine potently inhibits CYP1B1 activity, whereas CYP1A2 exhibits resistance to the inhibition (Lo et al., 2015). This led us to speculate that NC may induce CYP1 enzyme inhibition.
With the rapidly growing global interest in the use of natural products as dietary supplements and medical remedies, rigorous interdisciplinary studies on the efficacy, potential toxicity, and health benefits of these botanicals have become increasingly and considerably significant. In the present study, we investigated the interactions of NC with human recombinant CYP1 enzymes and explored the alteration of E2 metabolism mediated by NC.
Materials and Methods
Chemicals and Materials.
Nitidine chloride was used in the study with purity >98%, which was acquired from Chengdu Push Bio-Technology Co., Ltd. (Chengdu, China). Human recombinant CYP1A2 and CYP1B1 were obtained from BD Gentest (Woburn, MA). Human recombinant CYP1A1 was purchased from Cypex (Dundee, UK). Ethoxyresorufin (ERF), resorufin, E2, ethinyloestradiol (EE2), dansyl chloride, α-naphthoflavone (α-NF), and propranolol were supplied by Aladdin Industrial Corporation (Shanghai, China). NADPH was acquired from Sigma-Aldrich (St. Louis, MO). 2-OHE2 and 4-OHE2 were purchased from Toronto Research Chemicals Inc. (Toronto, Canada).
Determination of IC50 Values of NC-Mediated Inhibition of CYP1A1, 1B1, and 1A2.
Ethoxyresorufin O-deethylation (EROD) assays (Scheme 1) were performed with CYP1 enzymes to measure enzyme inhibition, according to a published method (Henderson et al., 2000). Briefly, CYP1A1, CYP1B1, or CYP1A2 (5.0 nM) was incubated with ERF (2.0 μM) in 200 μl of 100 mM potassium phosphate buffer (pH 7.4) containing NC (0–100 μM) and MgCl2 (3.2 mM). The incubations were started by adding NADPH (1.0 mM). In addition, α-NF, a known selective inhibitor of CYP1 enzymes (Shimada, 2017), was incubated with ERF as described previously to ensure the involvement of CYP1 enzymes in EROD metabolism. After 10-minute incubation at 37°C, the reaction mixtures were quenched with 200 μl ice-cold acetonitrile consisting of 50 ng/ml propranolol [internal standard (IS)], and then centrifuged at 19,000g for 10 minutes to remove protein. The supernatants were analyzed by the liquid chromatography–tandem mass spectrometry (LC-MS/MS) system as will be described subsequently. The formation of de-ethylation metabolite was linear for the first 10 minutes, and the concentrations of NC and α-NF required for 50% inhibition of catalytic activity (IC50 values) were calculated by curve fitting.
Determination of Kinetic Values for NC-Induced Inhibition of CYP1A1 and CYP1B1.
The kinetic values for NC-induced inhibition of CYP1 enzymes were determined by EROD assays. ERF (0.05–5 μM) was mixed with CYP1A1 or CYP1B1 (5.0 nM) in the presence of NC at concentrations of 0, 0.1, 0.5, or 2.5 μM. The same procedure was carried out for reaction initiation, reaction termination, and sample preparation as described previously. EROD activity was measured by LC-MS/MS–based monitoring of resorufin formation as follows. Enzyme activities were assessed by an established resorufin standard curve. Specifically, calibration standards were obtained by serially diluting resorufin stock solutions with PBS to final concentrations of 0.5, 1.0, 2.0, 5.0, 25, 100, and 500 nM, followed by adding an equal volume of ice-cold acetonitrile with 50 ng/ml propranolol (IS). Seven-point calibration curves were estimated by the plot of the response ratios of analytes to the IS (resorufin) against the resorufin concentrations, and the linearity was verified by applying the 1/x-weighted linear regression method.
Determination of Time- and NADPH-Dependent Inhibitory Effects of NC on CYP1A1 and CYP1B1.
Recombinant CYP1A1 or CYP1B1 (50 nM), NC (0 and 100 μM), and MgCl2 (3.2 mM) were mixed in 120 μl PBS, followed by addition of NADPH (1.0 mM) for the initiation of the reaction. After incubation at 37°C for 0, 5, 15, and 30 minutes, aliquots (20 μl) were withdrawn and transferred to secondary incubation mixtures containing ERF (2.0 μM), MgCl2 (2.0 mM), and NADPH (0.5 mM). The resulting mixture was incubated at 37°C for 10 minutes. In a separate study, similar microsomal incubation without NADPH was carried out to determine the effect of NADPH on enzyme inhibition. The reactions were terminated by adding an equal volume of ice-cold acetonitrile that contained propranolol as the IS. After deproteination by centrifuging, the resulting supernatants were submitted to LC-MS/MS analysis for assessment of residual catalytic activity.
Determination of IC50 Values of NC-Induced Inhibition of 2- and 4-Hydroxylation of E2 Mediated by CYP1 Enzymes.
The incubation mixtures were composed of E2 (20 μM), NC (0–100 μM), and MgCl2 (3.2 mM) fortified with CYP1A1, CYP1B1, or CYP1A2 (10 nM) in 100 μl of potassium phosphate buffer (100 mM, pH 7.4) containing 0.1% ascorbic acid. The enzymatic reactions were initiated by addition of NADPH (1.0 mM), followed by incubation at 37°C for 10 minutes. Reactions were stopped by mixing 500 μl of ethyl acetate with EE2 (IS, 25 nM). After vortexing for 30 seconds, the organic layer was collected and concentrated under a stream of nitrogen gas, and then the residues were dissolved in 100 μl of sodium bicarbonate buffer (0.1 M, pH 9.0) and 100 μl dansyl chloride (1.0 mg/ml in acetone). The solution was incubated at 60°C for 20 minutes. The resulting samples were cooled on ice and centrifuged at 19,000g for 10 minutes. The supernatants were injected into the LC-MS/MS system for analysis as subsequently described.
Determination of Kinetic Parameters for NC-Induced Inhibition of E2 Hydroxylation Mediated by CYP1A1.
The incubations containing CYP1A1 (10 nM) and E2 (0.5–80 μM) were performed with NC at concentrations of 0, 0.05, 0.1, 0.2, or 0.5 μM and NADPH (1.0 mM) at 37°C for 10 minutes. The resulting incubation mixtures and metabolite standards (2-OHE2 and 4-OHE2) were extracted and derivatized following the same procedure as previously described. After derivatization, all samples were analyzed by LC-MS/MS.
Cell Culture.
MCF-7 cells were generously provided by Dr. F.J. Zhang (Shenyang Pharmaceutical University). Cells were cultured in Dulbecco’s modified Eagle’s medium (HyClone; GE Healthcare Life Sciences, China) fortified with 10% FBS (Gibco, Australia) and 1% penicillin-streptomycin solution (HyClone; GE Healthcare Life Sciences) at 37°C in a 5% CO2 environment.
Evaluation of NC-Induced Inhibition of 2- and 4-Hydroxylation of E2 Mediated by CYP1 Enzymes in MCF-7 Cells.
MCF-7 cells were seeded on 24-well plates at a density of 1.2 × 105 cells per well. Following 24 hours of stabilization, cells were treated with Dulbecco’s modified Eagle’s medium containing NC (5.0, 10, and 20 μM). DMSO (0.4% v/v) was used as a negative control. After incubating for 1 hour, culture media were removed. The cells were washed once with PBS, followed by addition of E2 (1.0 μM) and 24-hour incubation. Cell media (1.0 ml) were then collected and extracted with ethyl acetate (2 × 2 ml) containing EE2 (IS, 5.0 nM) and 0.1% ascorbic acid. The organic layers were combined and dried under a gentle flow of nitrogen. The residues were derivatized as described previously, and the resultant samples were submitted to LC-MS/MS for analysis.
Simulation of Binding of NC with CYP1 Enzymes by Computer Modeling and Docking.
The LibDock module with the LibDockScore scoring function of Discovery Studio version 3.0 was employed to perform the computer modeling of NC. The docking of NC to putative active sites of three CYP1 enzymes was generated using crystallographic structures of CYP1A2 (Protein Data Bank Identification: 2HI4), CYP1A1 (Protein Data Bank Identification: 4I8V), and CYP1B1 (Protein Data Bank Identification: 3 PM0). During the docking process, the top 10 conformations were saved for each ligand based on the dock score value after energy minimization using the smart minimize method through the LibDock program. Finally, docking models of NC with CYP1 members were displayed by the PyMOL software program (http://www.pymol.org).
LC-MS/MS Analysis Method.
NC-mediated inhibition of EROD and E2 hydroxylation activity catalyzed by CYP1 enzymes was assessed by monitoring the generation of de-ethylation and hydroxylation metabolites, respectively. The products were analyzed on a 5500 triple quadrupole mass spectrometer from AB Sciex (Applied Biosystems, Foster City, CA) interfaced online with an Agilent 1260 Series HPLC (Agilent Technologies, Santa Clara, CA). Chromatographic separation was achieved on an analytical Promosil C18 column (100 × 4.6 mm, 5.0 μm; Agela Technologies, Inc., Tianjin, China) at the temperature of 25°C. Gradient elution was performed for resorufin with a flow rate of 0.8 ml/min, including mobile phase A (acetonitrile with 0.1% formic acid) and mobile phase B (water with 0.1% formic acid). The high-performance liquid chromatography gradient was set as follows: 10% A at 0–2.0 minutes; 10%–40% A at 2.0–2.5 minutes; 40%–95% A at 2.5–6.0 minutes; 95% A at 6.0–9.0 minutes; 95%–10% A at 9.0–9.5 minutes; and 10% A at 9.5–11.0 minutes. Isocratic elution was conducted for derivatized 2-OHE2 and 4-OHE2 with 88% (A) at a 1.0 ml/min flow rate. The injection volume was 5.0 μl. Quantification was conducted with an electrospray ionization source in positive mode. Multiple reactions monitoring scanning was used for targeted analysis with the ion transition (corresponding to collision energy and declustering potential) mass-to-charge ratio (m/z) 214.0 →186.0 for metabolite resorufin (35, 70), m/z 755.0 →521.0 for derivatized metabolites 2-OHE2 and 4-OHE2 (44, 50), m/z 260.3 →116.3 for IS propranolol (44, 77), and m/z 530.0 →171.0 for derivatized IS EE2 (35, 30), respectively. All operations and data analysis were processed by the Analyst software program (version 1.6.3; Applied Biosystems).
Data Analysis and Statistics.
To evaluate NC-induced inhibition of CYP1A1 and 1B1 and alteration of E2 metabolism, kinetic analyses were conducted following the Michaelis-Menten kinetic property. Velocity-substrate concentration (V-S) curves were fitted by no-weighting nonlinear least-squares regression, according to the Michaelis-Menten equation in the GraphPad Prism 5 software program (GraphPad Co. Ltd., San Diego, CA). The data were expressed as the mean ± S.D. of three independent experiments. For different types of inhibition, the kinetic values (KI and Ki) were calculated as follows: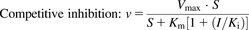
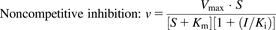
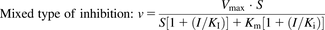 where I and Vmax are the NC concentration and maximal velocity, respectively; Km is the substrate concentration at one-half the Vmax value of the reaction; and Ki and KI are the inhibition constants for the binding of an inhibitor to the enzyme and enzyme-substrate complex, respectively.
where I and Vmax are the NC concentration and maximal velocity, respectively; Km is the substrate concentration at one-half the Vmax value of the reaction; and Ki and KI are the inhibition constants for the binding of an inhibitor to the enzyme and enzyme-substrate complex, respectively.
Results
NC-Induced Inhibition of CYP1 Enzymes.
EROD assays with recombinant enzymes were conducted to investigate NC-induced inhibitory effects on CYP1 enzymes. Among the three isozymes, NC was found to cause potent inhibition of CYP1A1 and CYP1B1 activities in a concentration-dependent manner with IC50 values of 0.28 ± 0.06 and 0.32 ± 0.02 μM, respectively (Table 1). However, the inhibitory efficiency of NC on CYP1A2 (IC50 > 50 μM) was considerably lower than that with CYP1A1 or CYP1B1 (Fig. 1). The IC50 values obtained from inhibition tests by α-NF are also listed in Table 1. These data indicate that NC selectively inhibited CYP1A1 and CYP1B1 activities, especially CYP1A1 activity.
NC- and α-NF–induced inhibition of EROD activity of CYP1A1, CYP1B1, and CYP1A2
Data represent the mean ± S.D. (n = 3).

NC-Induced inhibition of EROD activity catalyzed by CYP1 enzymes. CYP1A1, CYP1B1, or CYP1A2 (5 nM) was incubated with ERF (2.0 μM) and NC (0–100 μM) at 37°C for 10 minutes, followed by assessment of remaining enzyme activity. Data represent the mean ± S.D. (n = 3).
Time- and NADPH-Dependent Inhibition of NC on CYP1A1 and CYP1B1.
We initially probed different preincubation times on the inhibitory effects of CYP1A1 and CYP1B1 activities mediated by NC. As shown in Fig. 2, NC showed no time-dependent inhibition on CYP1A1 and 1B1. We also tested whether the presence of NADPH is required for enzymatic inactivation induced by NC. No significant difference in CYP1A1 and CYP1B1 activities was found after 30-minute microsomal incubation with or without NADPH. These results suggest that NC is a reversible inhibitor of CYP1A1 and CYP1B1.

Time- and NADPH-dependent inhibition of CYP1A1 (A) and CYP1B1 (B) by NC. CYP1A1 or CYP1B1 (50 nM) was incubated with vehicle (●) and NC (100 μM) in the presence (▲) or absence (■) of NADPH at 37°C for 0, 5, 15, and 30 minutes. Aliquots of the incubation mixtures were transferred to secondary incubation mixtures for determination of residual enzymatic activity.
Kinetic Analysis of NC-Induced Inhibition on CYP1A1 and CYP1B1.
According to the aforementioned IC50 results, CYP1A2-mediated EROD activity was hardly inhibited by NC. Thus, kinetic studies were carried out to further evaluate NC-induced inhibition on CYP1A1 and CYP1B1. Nonlinear regression analysis for competitive or mixed inhibition using GraphPad Prism 5 was used to determine the apparent Ki values and the mode of inhibition. The correlation coefficients (r) were 0.968–0.995. CYP1A1 catalyzed EROD activity with Vmax and Km values of 21.0 ± 2.3 nmol/min per nanomoles P450 and 1.30 ± 0.36 μM, respectively. CYP1B1 catalyzed EROD activity with Vmax and Km values of 5.71 ± 0.39 nmol/min per nanomoles P450 and 0.31 ± 0.03 μM, respectively. NC competitively inhibited CYP1B1 activity with a Ki value of 0.47 ± 0.05 μM. However, NC produced a mixed type of inhibition of CYP1A1 with Ki and KI values of 0.14 ± 0.04 and 0.19 ± 0.09 μM, respectively (Fig. 3; Table 2). The kinetic value (Ki) of CYP1B1 inhibition by NC was higher than that of CYP1A1 (Ki and KI), which is in accordance with the IC50 results obtained.

Kinetic analysis of NC-induced inhibition of EROD activity in a human recombinant CYP1A1 or CYP1B1 system. (A and C) show the velocity (v) vs. ERF concentration (S) plots of activity in the presence of NC at various concentrations as indicated. (B and D) show the Lineweaver-Burk plots for NC-induced inhibition of EROD activity mediated by CYP1A1 and CYP1B1. Data represent the mean ± S.D. (n = 3).
Kinetic parameters of NC-induced inhibition of EROD activity of CYP1A1, CYP1B1, and CYP1A2
Data represent the mean ± S.D. (n = 3).
Molecular Docking of NC with CYP1A1, 1A2, and 1B1.
To assist the explanation for the observed inhibition of CYP1A1 and 1B1, computer modeling of the binding of NC to CYP1 enzymes was performed to compare NC affinity with the three CYP1 enzymes and to define the interactions with the crucial amino acid residue(s). LibDock was used to perform the interplay of NC with CYP1 enzymes (Fig. 4). The dockings of NC to the active sites of CYP1 enzymes showed LibDockScore values in the order of CYP1A1 (153.191) > CYP1B1 (151.352) > CYP1A2 (120.382). The binding of NC to CYP1A1 displayed a higher score with a lower IC50 value in activity inhibition (Table 1). The methylenedioxy ring of NC lay closely to the heme moiety of the three CYP1 members. Ser127 of CYP1B1 and Ser116 of CYP1A1 were found to interact with one of the methoxy moieties of NC via hydrogen bonds, respectively. However, no hydrogen bond formation was found between the methoxy moieties of NC and CYP1A2. Instead, two unfavorable interaction bonds were observed in CYP1A2. Other chemical interactions, including π-π, π-alkyl, etc., are presented in Fig. 4. There was no significant difference between CYP1 isozymes in forming hydrophobic interactions with NC. The docking results suggest that Ser127 of CYP1B1 and Ser116 of CYP1A1 might be of significance in NC-induced enzyme inhibition.

Three-dimensional structural docking of NC with CYP1A1 (A), CYP1B1 (B), and CYP1A2 (C). The nitrogen and oxygen atoms are shown in dark blue and red, respectively. The heme prosthetic group of P450 enzymes is shown in magenta with iron in dark red. The hydrogen bond formation between NC and the amino acid residues of apoprotein of CYP1 enzymes is shown by yellow dashed lines.
NC-Induced Inhibition of 2- and 4-Hydroxylation of E2 Mediated by CYP1 Enzymes.
Coincubation studies were performed to determine the inhibitory effects of NC on CYP1 enzyme–mediated E2 hydroxylation. NC showed prominent inhibition on CYP1A1-catalyzed 2- and 4-hydroxylation of E2 with IC50 values of 0.16 ± 0.004 and 0.25 ± 0.04 μM, respectively, which were significantly lower than the values for CYP1A2 (IC50 = 13.2 ± 5.56 and 5.35 ± 0.62 μM) and CYP1B1 (IC50 = 35.0 ± 6.44 and 8.27 ± 2.40 μM) (Fig. 5; Table 3). Furthermore, the IC50 value for the inhibition of 2-hydroxylation activity of CYP1A1 was about 60% of that for 4-hydroxylation, revealing that NC preferentially inhibited CYP1A1-mediated E2 2-hydroxylation activity.

NC-induced inhibition of E2 2-hydroxylation (A) and 4-hydroxylation (B) activities catalyzed by CYP1 enzymes. CYP1A1, CYP1B1, or CYP1A2 (10 nM) was incubated with E2 (20 μM) and NC (0–100 μM), followed by assessment of remaining enzyme activity. Data represent the mean ± S.D. (n = 3).
NC-Induced inhibition of E2 hydroxylation activity of CYP1A1, CYP1B1, and CYP1A2
Data represent the mean ± S.D. (n = 3).
Kinetic Parameters for NC-Induced Inhibition of E2 Hydroxylation Mediated by CYP1A1.
Due to the strong inhibition of CYP1A1-mediated 2- and 4-hydroxylation of E2 by NC, kinetic analyses were conducted with various concentrations of NC in the CYP1A1 system to characterize the type of reversible inhibition. The plots of the velocity versus substrate concentrations in CYP1A1-catalyzed 2- and 4-hydroxylation of E2 showed hyperbolicity (Fig. 6, A and C). The Km and Vmax values for 2-hydroxylation of E2 by CYP1A1, 1B1, and 1A2 were 3.38 ± 0.69, 2.26 ± 0.30, and 23.7 ± 4.06 μM, and 6.91 ± 0.33, 0.45 ± 0.04, and 50.6 ± 4.93 nmol/min per nanomoles P450, respectively. The Km and Vmax values for 4-hydroxylation of E2 by CYP1A1, 1B1, and 1A2 were 28.9 ± 11.5, 0.87 ± 0.16, and 24.8 ± 4.78 μM, and 0.22 ± 0.04, 1.21 ± 0.12, and 0.97 ± 0.14 nmol/min per nanomoles P450, respectively (Table 3). The catalytic efficiency (Vmax/Km) of CYP1 enzyme–mediated 2- and 4-hydroxylation of E2 were 2.04 and 0.008 ml/min per nanomoles P450 (CYP1A1), 0.20 and 1.39 ml/min per nanomoles P450 (CYP1B1), and 2.14 and 0.039 ml/min per nanomoles P450 (CYP1A2), respectively (Table 3). The calculated activity ratio (2-/4-hydroxylation) of CYP1A1, 1B1, and 1A2 were 255, 0.14, and 55, respectively, indicating that CYP1A1 and 1A2 primarily catalyzed the formation of 2-OHE2, and CYP1B1 primarily catalyzed the formation of 4-OHE2 (Scheme 2; Table 3). Lineweaver-Burk plots showed that NC-induced inhibition of E2 2-hydroxylation activity mediated by CYP1A1 was best fit to a mixed type of inhibition with Ki and KI values of 0.052 ± 0.007 and 0.210 ± 0.016 μM (Fig. 6B; Table 4). E2 4-hydroxylation activity inhibition by NC was fit well to a competitive inhibition model with a Ki value of 0.26 ± 0.03 μM (Fig. 6D; Table 4).

Proposed effect of NC on E2 chemical carcinogenesis. CYP1A1 and 1A2 primarily catalyze the formation of 2-OHE2 (detoxification biomarker). CYP1B1 primarily catalyzes the formation of 4-OHE2 (genotoxic biomarker), which is further oxidized to genotoxic estradiol-3,4-quinone (3,4-E2-Q). NC has been reportedly demonstrated to markedly decrease 2-hydroxylation and relatively increase 4-hydroxylation metabolism of E2, as shown by the green and red arrows.

Kinetic analysis of NC-induced inhibition of E2 hydroxylation activity in a human recombinant CYP1A1 system. (A and C) show the velocity (v) vs. E2 concentration (S) plots of activity in the presence of NC at various concentrations as indicated. (B and D) show the Lineweaver-Burk plots for NC-induced inhibition of E2 hydroxylation activity mediated by CYP1A1. Data represent the mean ± S.D. (n = 3).
Kinetic parameters of NC-induced inhibition of E2 hydroxylation activity of CYP1A1, CYP1B1, and CYP1A2
Data represent the mean ± S.D. (n = 3).
Effects of NC on the Formation of E2-Hydroxylation Metabolites Mediated by CYP1 Enzymes in MCF-7 Cells.
As further investigation, the potential inhibitory effects of NC on CYP1A1 and 1B1 were evaluated in MCF-7 cells. As shown in Fig. 7, both 2- and 4-OHE2 were detected in cell media after E2 (1.0 μM) treatment with or without NC. The relative amounts of 2- and 4-OHE2 generated in vehicle- and NC-treated groups were estimated. As expected, NC elicited inhibitory effects on 2- and 4-hydroxylation of E2 in a concentration-dependent manner. Additionally, significantly decreased levels of 2-OHE2 were found after cells were treated with NC at concentrations of 10 μM (P < 0.05) or 20 μM (P < 0.01). The results suggest that NC had a stronger inhibition effect on the formation of nontoxic 2-OHE2 in MCF-7 cells.

Effect of NC on the formation of E2-hydroxylation metabolites (2- and 4-OHE2) in MCF-7 cells. Data represent the mean ± S.D. (n = 3).
Discussion
Botanical dietary supplements consisting of a variety of chemopreventive constituents with many pharmacological activities are popularly used as natural alternatives to medicines. However, drug-drug interactions are frequently caused by some natural product–mediated P450 enzyme inhibition, possibly resulting in severe adverse effects (Danton et al., 2013). P450 inhibition can be classified into reversible and irreversible inhibition (VandenBrink and Isoherranen, 2010). P450s are important phase I enzymes responsible for various biotransformations of endogenous substrates and xenobiotics, not only existing in intrahepatic tissues but also extensively distributed across extrahepatic tissues (Vrba et al., 2004; Liu et al., 2017). Thus, safe consumption of such natural products is becoming a public concern. NC is the major component in Zanthoxylum nitidum (Roxb.) DC widely used in traditional Chinese medicine. Our previous research demonstrated that NC shows no remarkable time-dependent inhibition on other major P450 enzymes in liver except for CYP2D6 (Mao et al., 2018). Carbene intermediate produced in the metabolic pathway of the methylenedioxyphenyl group of NC might lead to the quasi-irreversible inactivation of CYP2D6. In the present study, we examined the inhibitory effects of NC on three CYP1 isozymes that are considered to be linked with the generation of carcinogens. The majority of CYP1 inhibitors contain two or more fused aromatics, exhibiting molecular planarity. Heterocyclic rings and nitrogen frequently occur in such inhibitors as part of the ring system and/or in a substituent group (Zanger and Schwab, 2013; Lee et al., 2016). NC is such a polyaromatic hydrocarbon compound containing a nitrogen heterocycle. Our findings revealed that NC efficiently inhibited EROD activities mediated by CYP1A1 and CYP1B1 in a concentration-dependent manner with IC50 values of 0.28 and 0.32 μM, respectively, but CYP1A2 was resistant to the inhibition by NC (Fig. 1; Table 1). However, time- and NADPH-dependent inhibition of CYP1 enzymes by NC was not observed under the present conditions (Fig. 2), indicating that NC is a reversible inhibitor of CYP1A1 and 1B1. Further kinetic studies clearly demonstrated that NC exhibited competitive inhibition of CYP1B1 with a Ki value of 0.47 μM, suggesting that NC bound to the same pocket of CYP1B1 enzyme as that for ERF (Fig. 3; Table 2). NC showed the a mixed type of inhibition for CYP1A1 with lower Ki and KI values of 0.14 and 0.19 μM, respectively, which suggests that NC does not only share the identical binding pocket with the probe substrate (ERF) but can also bind to a different pocket of the CYP1A1 enzyme. This observation indicates that NC demonstrated more significant inhibitory effects for CYP1A1 than for CYP1B1. In addition, molecular modeling of NC with CYP1 enzymes was conducted to help clarify the NC-induced difference in inhibition of the three enzymes. Hydrogen-bonding and π-π interactions were proposed to play a critical role in the binding of alkoxy derivatives of heterocyclic compounds to CYP1 enzymes (Lo et al., 2013). NC was found to interact with amino acid residue Ser127 of CYP1B1 or Ser116 of CYP1A1 via hydrogen-bonding interactions, respectively, but no such hydrogen bond was generated in CYP1A2 (Fig. 4). The LibDockScore results also showed that the affinities of NC with CYP1A1 and 1B1 were higher than that of CYP1A2. Based on the docking score and molecular interactions with active site residues, the hydrogen bonds of CYP1A1 and CYP1B1 formed with NC might increase the inhibitory susceptibility of the two enzymes.
E2 is one of the major estrogens in women and has been the standard treatment option for postmenopausal symptom relief for decades (Wang et al., 2016). However, it is well known that prolonged exposure of estrogens plays a role in the breast cancer etiology, especially in postmenopausal women (Takemura et al., 2010). As in most other countries, breast cancer is now the most common cancer in Chinese women; cases in China account for 9.6% of all deaths from breast cancer worldwide (Fan et al., 2014). CYP1A1, 1B1, and 1A2 were demonstrated to mainly catalyze the oxidation of E2 to 2-OHE2 and 4-OHE2 (Lee et al., 2003). The 2-hydroxylation pathway negatively correlates with breast cancer risk, whereas the 4-hydroxylation pathway is likely an important initiator and promotor in breast cancer (Dunlap et al., 2017). 4-OHE2 is biotransformed to 3,4-E2-quinone, a known electrophilic species, causing the formation of DNA adducts and tumorigenesis (Yager, 2015; Wang et al., 2016). Reactive oxygen species can be also produced in this metabolic pathway, contributing to oxidative DNA damage. 2-Methoxyestradiol, generated from the metabolism of catechol by catechol-O-methyltransferase, blocks the further oxidation of 2-OHE2. Meanwhile, 2-methoxyestradiol, an inhibitor of CYP1B1, which is responsible for the 4-hydroxylation pathway, has been recently considered to be a promising agent to fight metastatic breast cancer (Schwarz et al., 2011). Thus, 2-OHE2 and 4-OHE2 may be regarded as detoxification and genotoxic biomarkers, respectively (Scheme 2). Our results showed that the formation of 2-OHE2 was primarily mediated by CYP1A1 and CYP1A2, and the formation of 4-OHE2 was primarily mediated by CYP1B1 (Scheme 2), which is consistent with a previous report (Itoh et al., 2010). The catalytic efficiency of E2 2-hydroxylation and 4-hydroxylation by CYP1A1 is approximately 255-fold higher than that of CYP1A2 (55-fold) and CYP1B1 (0.14-fold) (Table 3). Nevertheless, the IC50 value for NC-induced inhibition of E2 2-hydroxylation by CYP1A1 was about 60% of the value of 4-hydroxylation, indicating that NC is more potent in inhibiting the generation of 2-OHE2 (Table 3). Kinetic analysis displayed that NC is a mixed type of inhibitor of E2 2-hydroxylation with Ki and KI values of 0.052 and 0.21 μM, and a competitive inhibitor of E2 4-hydroxylation with a Ki value of 0.26 μM (Fig. 6; Table 4). No significant inhibition of E2 metabolism induced by NC was observed in CYP1B1 or CYP1A2 incubation systems. Additionally, the MCF-7 cell model study showed that NC significantly decreased the formation of 2-OHE2 (Fig. 7). Given together, the effect of NC on E2 chemical carcinogenesis is proposed as shown in Scheme 2. These data suggest that the NC-induced inhibitory effect on E2 oxidative metabolism in vitro was mainly contributed by the CYP1A1-mediated 2-hydroxylation pathway. The reduced production of 2-methoxyestradiol may relatively result in the accumulation of 4-OHE2. Thus, potential drug-drug interactions should be considered in the dosage regimen of NC for chemoprotection against the toxicity of 4-OHE2.
In conclusion, NC is a mixed type of inhibitor of CYP1A1 and a competitive inhibitor of CYP1B1. The observed preferential inhibition of NC on CYP1A1-mediated E2 2-hydroxylation suggests that 4-OHE2-mediated genotoxicity might be increased by NC.
Authorship Contributions
Participated in research design: Mao, Zheng, Peng.
Conducted experiments: Mao, Q. Wang, Yang, Li, Lin.
Performed data analysis: Mao, J. Wang.
Wrote or contributed to the writing of the manuscript: Mao, Zheng.
Footnotes
- Received February 21, 2019.
- Accepted May 24, 2019.
↵1 J.Z. and Y.P. contributed equally to this work.
The authors declare that there are no conflicts of interest.
This work was supported in part by the National Natural Science Foundation of China [Grants 81830104, 81430086, 81773813, and U1812403].
Abbreviations
- E2
- estradiol
- EE2
- ethinyloestradiol
- ERF
- ethoxyresorufin
- EROD
- ethoxyresorufin O-deethylation
- IS
- internal standard
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- m/z
- mass-to-charge ratio
- NC
- nitidine chloride
- α-NF
- α-naphthoflavone
- 2-OHE2
- 2-hydroxyestradiol
- 4-OHE2
- 4-hydroxyestradiol
- P450
- cytochrome P450
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}