


Abstract
The drug-metabolizing enzymes that contribute to the metabolism or bioactivation of a drug play a crucial role in defining the absorption, distribution, metabolism, and excretion properties of that drug. Although the overall effect of the cytochrome P450 (P450) family of drug-metabolizing enzymes in this capacity cannot be understated, advancements in the field of non-P450–mediated metabolism have garnered increasing attention in recent years. This is perhaps a direct result of our ability to systematically avoid P450 liabilities by introducing chemical moieties that are not susceptible to P450 metabolism but, as a result, may introduce key pharmacophores for other drug-metabolizing enzymes. Furthermore, the effects of both P450 and non-P450 metabolism at a drug’s site of therapeutic action have also been subject to increased scrutiny. To this end, this Special Section on Emerging Novel Enzyme Pathways in Drug Metabolism will highlight a number of advancements that have recently been reported. The included articles support the important role of non-P450 enzymes in the clearance pathways of U.S. Food and Drug Administration–approved drugs over the past 10 years. Specific examples will detail recent reports of aldehyde oxidase, flavin-containing monooxygenase, and other non-P450 pathways that contribute to the metabolic, pharmacokinetic, or pharmacodynamic properties of xenobiotic compounds. Collectively, this series of articles provides additional support for the role of non-P450–mediated metabolic pathways that contribute to the absorption, distribution, metabolism, and excretion properties of current xenobiotics.
Introduction
The identification of drug metabolism pathways and their relative importance is a prominent aspect in the determination of the pharmacokinetic properties of most xenobiotics (Wienkers and Heath, 2005; Foti et al., 2012). The scientific basis of these studies resides in multiple disciplines, including physiology, enzymology, metabolic biotransformation, in silico modeling and toxicology, among others (Lee et al., 2003). As such, the characterization of the enzymes involved in the metabolism of a new compound, the determination of its metabolic stability and primary biotransformation pathways, and the ability of a compound to inhibit or induce drug-metabolizing enzymes are all major facets of the drug discovery and development continuum. Generically speaking, drug metabolism can be thought of as the biologic conversion of hydrophobic, drug-like molecules into more polar metabolites that are then subject to facile excretion. In turn, the plasma and extravascular exposure of the parent drug and subsequent metabolites is determined, thus defining the safety and efficacy profile of the drug. Given the defining role of drug metabolism in safety and efficacy, it becomes no surprise that regulatory agencies emphasize the importance of characterizing the drug metabolism profile in the development process for new investigational drugs (Bohnert et al., 2016).
The biotransformation reactions catalyzed by drug-metabolizing enzymes are generally grouped into phase I (involving oxidation, reduction, and hydrolysis) or phase II (conjugation-based) reactions (Williams, 1969). Transporter-based interactions are often referred to as phase III reactions; although these interactions are outside the scope of this special section, many thorough reviews are currently available on the topic (Giacomini et al., 2010; Keogh, 2012). Phase I reactions are commonly catalyzed by enzymes such as the cytochromes P450 (P450), flavin-containing monooxygenases (FMOs), aldehyde oxidases (AOs), carboxylesterases (CESs), epoxide hydrolases, alcohol and aldehyde dehydrogenases (ADHs and ALDHs, respectively), and ketoreductases, in addition to others (Rendic and Di Carlo, 1997; Satoh and Hosokawa, 2006; Cashman, 2008; Decker et al., 2009; Strolin Benedetti, 2011; Garattini and Terao, 2012). Drug-metabolizing enzymes responsible for the phase II conjugative pathways include the UDP-glucuronosyltransferases (UGTs), sulfotransferases, N-acetyltransferases, glutathione transferases, methyltransferases, and acyl-CoA synthetases. Examples of phase I and phase II enzymes, along with their respective cofactors, substrates, inhibitors, and tissue location, are shown in Tables 1 and 2 (Riddle and Jencks, 1971; Trager, 1989; Ghersi-Egea et al., 1993; Matsui and Homma, 1994; Riley and Hanzlik, 1994; Dutton, 1997; Rendic and Di Carlo, 1997; Burchell et al., 1998; Halpert et al., 1998; Burchell, 1999; Weinshilboum et al., 1999; Tukey and Strassburg, 2000; Dalvie et al., 2002; Ortiz de Montellano and De Voss, 2002; Coughtrie and Fisher, 2003; Ding and Kaminsky, 2003; Nelson and Trager, 2003; Guengerich, 2005; Satoh and Hosokawa, 2006; Cashman, 2008; Dourado et al., 2008; Nakamura et al., 2008; Decker et al., 2009; Remmel and Zhou, 2009; Strolin Benedetti, 2011; Fasinu et al., 2012; Foti and Fisher, 2012; Garattini and Terao, 2012). A number of comprehensive resources detailing the chemistry and reaction mechanisms for many of the enzymes are readily available (Trager, 1989; Riley and Hanzlik, 1994; Burchell et al., 1998; Halpert et al., 1998; Burchell, 1999; Dalvie et al., 2002; Nelson and Trager, 2003; Foti et al., 2012). This special section will aim to highlight the increasing emphasis placed on non-P450–catalyzed reactions, specifically those involved in oxidative metabolism.
Examples of cofactors, substrates, inhibitors, and primary tissue locations for P450 and UGT drug-metabolizing enzymes
Cofactors, substrates, inhibitors, and primary tissue locations for additional drug-metabolizing enzymes
An additional topic worthy of highlighting in a special section devoted to emerging and novel drug metabolism pathways is the recent advancements in mass spectrometry–based proteomics that have allowed the field to re-evaluate the expression levels of drug-metabolizing enzymes in the liver and other tissues. In the early years of characterizing expression patterns of drug-metabolizing enzymes, determination of P450 protein levels used selective antibodies, whereas the analysis of non-P450 phase I and phase II drug-metabolizing enzymes was primarily limited to measuring mRNA expression. Indeed, the field of proteomics has greatly expanded the tools available to characterize drug-metabolizing enzyme expression patterns (Ohtsuki et al., 2012; Schaefer et al., 2012). To highlight the magnitude of these advancements in recent years, we mined the literature for studies on the expression levels of drug-metabolizing enzymes that reported protein expression on a picomole/milligram basis. Reported methods included Western blot analyses as well as mass spectrometry–based approaches in various matrices, and the two analytical methods were weighted equally in terms of calculating the average expression levels for each enzyme. Averages and standard deviations were weighted based on the number of individual samples reported in each study. On the basis of an initial analysis of 25 literature reports, a composite view of drug-metabolizing enzyme expression is shown in Fig. 1; this allows for a statistically simplistic comparison of P450 versus non-P450 enzyme expression, as well as an assessment of the variability observed within the reported expression levels for each enzyme. It is quite likely that the overall variability results from a combination of pharmacogenetics, patient history, and choice of analytical methods, as discussed in recent literature reports (Tracy et al., 2016).
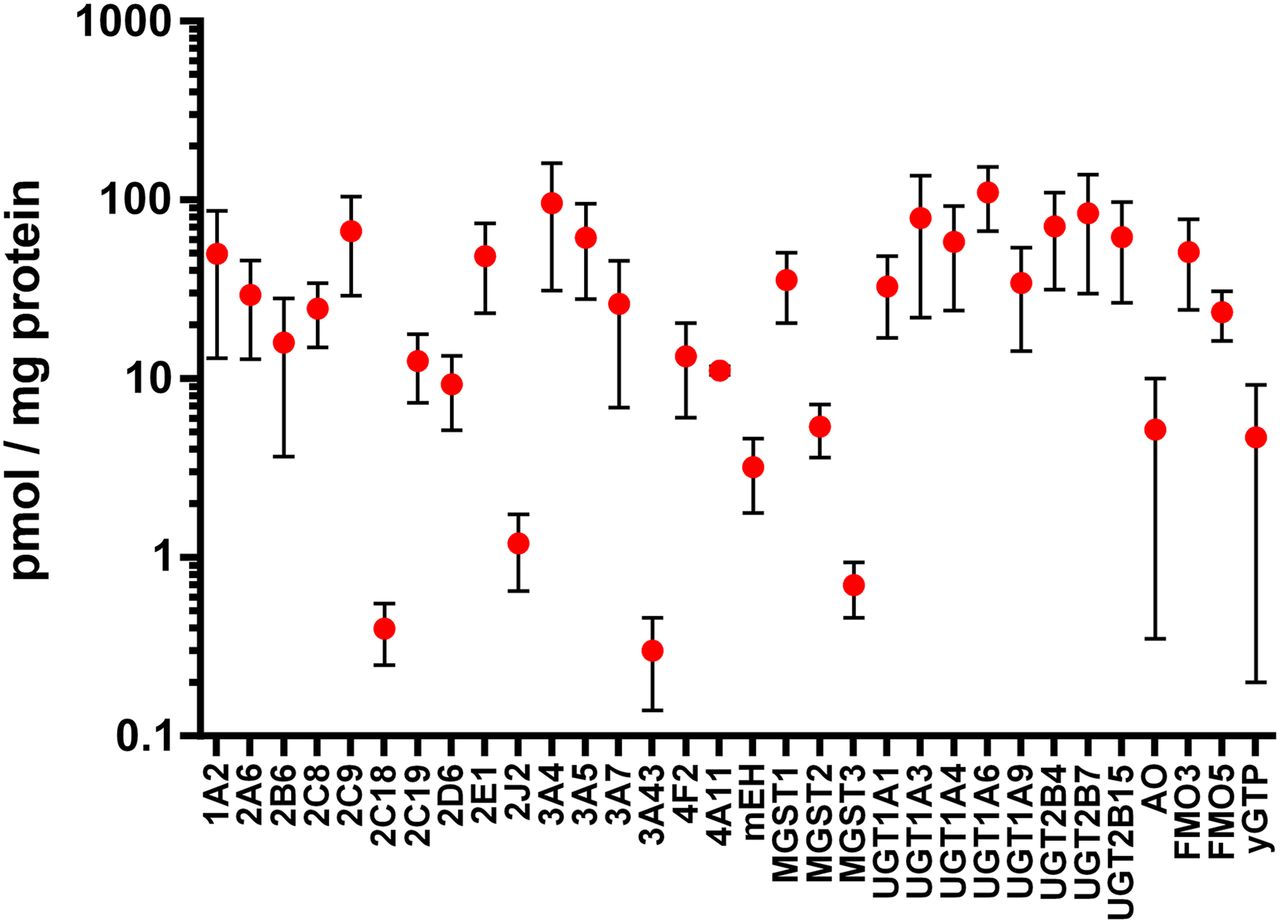
Estimated expression levels of hepatic drug-metabolizing enzymes as described in multiple literature reports. Protein data were included from both Western blot and mass spectrometry–based methods (Shimada et al., 1994; Overby et al., 1997; Richardson et al., 1997; Lasker et al., 1998; Zanger et al., 2001; Coller et al., 2002; Lin et al., 2002; Lamba et al., 2003; Westlind-Johnsson et al., 2003; Koukouritaki et al., 2004; Rettie and Jones, 2005; Hofmann et al., 2008; Langenfeld et al., 2009; Klein et al., 2010; Naraharisetti et al., 2010; Kawakami et al., 2011; Ohtsuki et al., 2012; Schaefer et al., 2012; Barr et al., 2013; Fu et al., 2013; Zanger and Schwab, 2013; Achour et al., 2014; Michaels and Wang, 2014; Song et al., 2015; Chen et al., 2016).
The overall aim of this special section is to briefly highlight the emerging contributions of both P450 and non-P450 drug-metabolizing enzymes, with a specific emphasis on the oxidative pathways catalyzed by the latter. Table 3 includes examples of the role of non-P450 enzymes in affecting both the pharmacokinetics and pharmacodynamics of various xenobiotics (using the University of Washington Drug Interaction Database, https://didb.druginteractioninfo.org), and a search of the recent literature supports these findings (Cerny, 2016). The increase in the identification of metabolic pathways catalyzed by non-P450 enzymes will be discussed, as will the role of non-P450 enzymes in phase I functionalization reactions. Novel aspects of phase II enzymes will also be presented. Ultimately, together with the articles included in this special section, a case will be made for the effect of these emerging pathways on pharmacokinetics, drug interactions, and safety and efficacy profiles of novel therapeutics.
Examples (nonexhaustive) of xenobiotic compounds from the University of Washington Drug Interaction Database whose pharmacokinetics or pharmacodynamics have been reported to be affected by non-P450 oxidative enzymes since 2010
Cytochrome P450 Enzymes, UDP-Glucuronosyltransferases, and Sulfotransferases
Although the main focus of this special section is on emerging metabolic pathways of a non-P450 nature, no commentary on drug-metabolizing enzymes would be complete without some mention of P450 enzymology. P450s are a superfamily of heme-containing enzymes that are responsible for the oxidation or reduction of the majority of drugs currently in use (Ortiz de Montellano and De Voss, 2002). Over 57 isoforms are reported to make up the family, although the number of isoforms directly involved in the metabolism of xenobiotics is more limited, with CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 being the P450 isoforms with significant roles in drug metabolism (Guengerich, 2005). The endoplasmic reticulum (ER) of the liver, intestine, lung, kidney, brain, and nasal mucosa cells is the primary site of P450 expression, with the active site of the P450 enzymes oriented toward the cytosolic side of the ER membrane (Ding and Kaminsky, 2003). The mechanism of the P450 reaction cycle necessitates the transfer of electrons from NADPH through various redox partners such as P450 reductase and cytochrome b5 (Iyanagi and Mason, 1973; Vermilion and Coon, 1978; Vermilion et al., 1981; Schenkman and Jansson, 1999). Although limited “novel metabolic pathways” catalyzed by P450 have been reported in recent years, efforts have focused on the characterization of minor isoforms such as CYP2J2 or the CYP4Fs as well as the contribution of P450 metabolism at the therapeutic site of action to the overall efficacy of a given drug (Kirischian and Wilson, 2012; Xu et al., 2013; Eksterowicz et al., 2014; Michaels and Wang, 2014; Foti et al., 2015; Uehara et al., 2015).
Similarly, UGTs and sulfotransferases represent other families of well studied drug-metabolizing enzymes. The UGTs catalyze the addition of glucuronic acid from uridine diphosphoglucuronic acid to a multitude of endogenous compounds and xenobiotics (Miners et al., 2004; Radominska-Pandya et al., 2005). The primary pharmacophore requirement is adequate nucleophilicity to accept transfer of the glucuronic acid moiety, with functional groups including aliphatic alcohols, carboxylic acids (resulting in acyl glucuronides), phenols, amines, and thiols (Tukey and Strassburg, 2000). Well characterized substrates include bilirubin, estradiol, serotonin, propofol, and morphine. Furthermore, as metabolite contributions to drug–drug interactions have gained importance since the issuance of new regulatory guidances, examples of metabolites formed by non-P450s that can contribute to clinically relevant drug–drug interactions have been identified (VandenBrink and Isoherranen, 2010; Yeung et al., 2011; Yu and Tweedie, 2013). A few classic examples exemplify metabolites formed by non-P450 enzymes in playing a role in inhibiting P450 are gemfibrozil (Backman et al., 2002; Wang et al., 2002), clopidogrel (Tornio et al., 2014), and, more recently, deleobuvir (Sane et al., 2016), in which UGT-catalyzed acyl glucuronide formation is responsible for inactivating CYP2C8 and resulting in significant drug–drug interactions.
Conversely, the addition of a sulfate group to a nucleophilic ligand is catalyzed by sulfotransferases. The enzymes use 3′-phosphoadenosine 5′-phosphosulfate as the sulfate donor; although the primary outcome of the sulfation reaction is a molecule with increased hydrophilicity and decreased pharmacological activity, a number of reports of sulfotransferase-catalyzed bioactivation have been noted, as in the case of 3-n-butylphthalide (Chou et al., 1995a,b, 1998; Gamage et al., 2006; Diao et al., 2014). The main pharmacophores for the sulfotransferases are aliphatic and aromatic hydroxyl groups, in addition to N-substituted hydroxylamines (Klaassen and Boles, 1997). In general, P450-, UGT-, and sulfotransferase-catalyzed metabolic pathways have been well covered in the literature and will not be discussed in detail in this collection of work on emerging pathways.
Flavin-Containing Monooxygenases
FMOs are involved in the metabolism of a wide array of xenobiotics, owing in part to the fact that a soft nucleophile such as a sulfur or nitrogen heteroatom is all that is necessary to convey the structural requirements to render the compound a substrate of FMOs (Ziegler, 1990). In addition to nucleophilicity, the size and charge state of a compound as well as the active site topography of the FMO active sites are important determinants of FMO-catalyzed metabolic profiles (Ziegler, 1990; Riley and Hanzlik, 1994; Rettie et al., 1995; Krueger et al., 2009). Although most readers will be familiar with FMO-catalyzed reactions such as benzydamine N-oxidation, recent efforts have focused on the role of FMOs in drug clearance, drug–drug interactions, and even in homeostatic roles such as the regulation of glucose and lipid metabolism and subsequent onset of atherosclerosis through the formation of metabolites such as trimethylamine-N-oxide (Bennett et al., 2013; Shih et al., 2015; Shimizu et al., 2015). Well known inhibitors of FMOs include indole-3-carbinol and methimazole. The role of FMOs in the conversion of a thiocarbonyl functionality to a reactive metabolite is well known (Ji et al., 2007).
Other examples in which FMO-mediated metabolism leads to formation of reactive metabolites include the oral antifungal agent ketoconazole and the synthetic neurotoxicant iminodipropionitrile (IDPN). Administration of ketoconazole has led to hepatic damage in the clinic on several occasions (Rodriguez and Buckholz, 2003). In addition, significant covalent binding is observed when [3H]-ketoconazole is incubated with NADPH-fortified rat liver microsomes, an observation that is abrogated after incubation of ketoconazole in heat-inactivated microsomes and suggests the involvement of FMOs. Deacetyl ketoconazole is the primary metabolite of ketoconazole and has been implicated in the hepatotoxicity of ketoconazole (Whitehouse et al., 1990). Rodriguez et al. proposed that deacetyl ketoconazole is converted to a secondary hydroxylamine metabolite, which in turn is oxidized to potentially reactive nitrone and/or dialdehyde metabolites (Fig. 2A) (Rodriguez and Acosta, 1997a; Rodriguez and Buckholz, 2003). Although there is no direct evidence that supports the formation or reactivity of the nitrone or dialdehyde intermediate, the proposal is consistent with the results from studies in rat hepatocyte culture systems that indicated the deacetyl ketoconazole metabolite to be more hepatotoxic than the parent (Rodriguez and Acosta, 1997b).

FMO-mediated bioactivation of secondary amines in ketoconazole (A) and IDPB (B) via a hydroxylamine intermediate.
The vestibular and auditory neurotoxicities that result after administration of IDPN are also attributed to FMO-catalyzed conversion of the compound to the putative N-hydroxy-3,3′-iminodipropionitrile (HOIDPN; Fig. 2B) (Jacobson et al., 1987; Morandi et al., 1987; Nace et al., 1997). This proposal is based on the fact that coadministration of methimazole, an FMO1/FMO3 inhibitor, with IDPN prevents these IDPN-mediated adverse events. Furthermore, neurotoxic events observed after administration of HOIDPN are 2- to 8-fold greater than those observed after administration of the parent compound (Crofton et al., 1996). It was suggested that one possible pathway could involve conversion of HOIDPN to cyanoacetaldehyde via the putative nitrone intermediate, which can transform protein-based amino groups to cyanoenamine adducts (Jacobson et al., 1987).
FMO has also been reported to play a major role in the conversion of the parent drug to active metabolites. Ethionamide, thiacetazone, albendazole, and fenbendazole represent examples of conversion of parent to an active metabolite by FMO. As noted above, the antituberculosis drug ethionamide is converted to ethionamide S-oxide by FMO, which is then further oxidized to the sulfinic acid derivative and ultimately to the 2-ethyl-4-carboxamidopyridine metabolite (Fig. 3A) (Vannelli et al., 2002). Assessment of biologic activity of the S-oxide metabolites showed that this metabolite retained full activity of ethionamide against Mycobacterium tuberculosis (Johnston et al., 1967). Similarly, human FMO1 and FMO3 have been shown to catalyze the oxidative activation of thiacetazone to isolable sulfinic acid and carbodiimide metabolites (Fig. 3B), the latter of which readily reacts with glutathione (Qian and Ortiz de Montellano, 2006). Albendazole and fenbendazole represent interesting examples that undergo enantioselective sulfoxidation of the prochiral sulfide moiety, yielding active sulfoxide metabolites albendazole sulfoxide (Hennessy et al., 1989) and fenbendazole sulfoxide or oxfendazole (Fig. 3C) (Marriner and Bogan, 1981). Although the involvement of both P450 and FMO systems in the sulfoxidation has been demonstrated in various species including humans, FMO specifically catalyzed the formation of (+)-albendazole sulfoxide and oxfendazole, whereas the P450 catalyzed the formation of the (−)-enantiomer (Virkel et al., 2004).
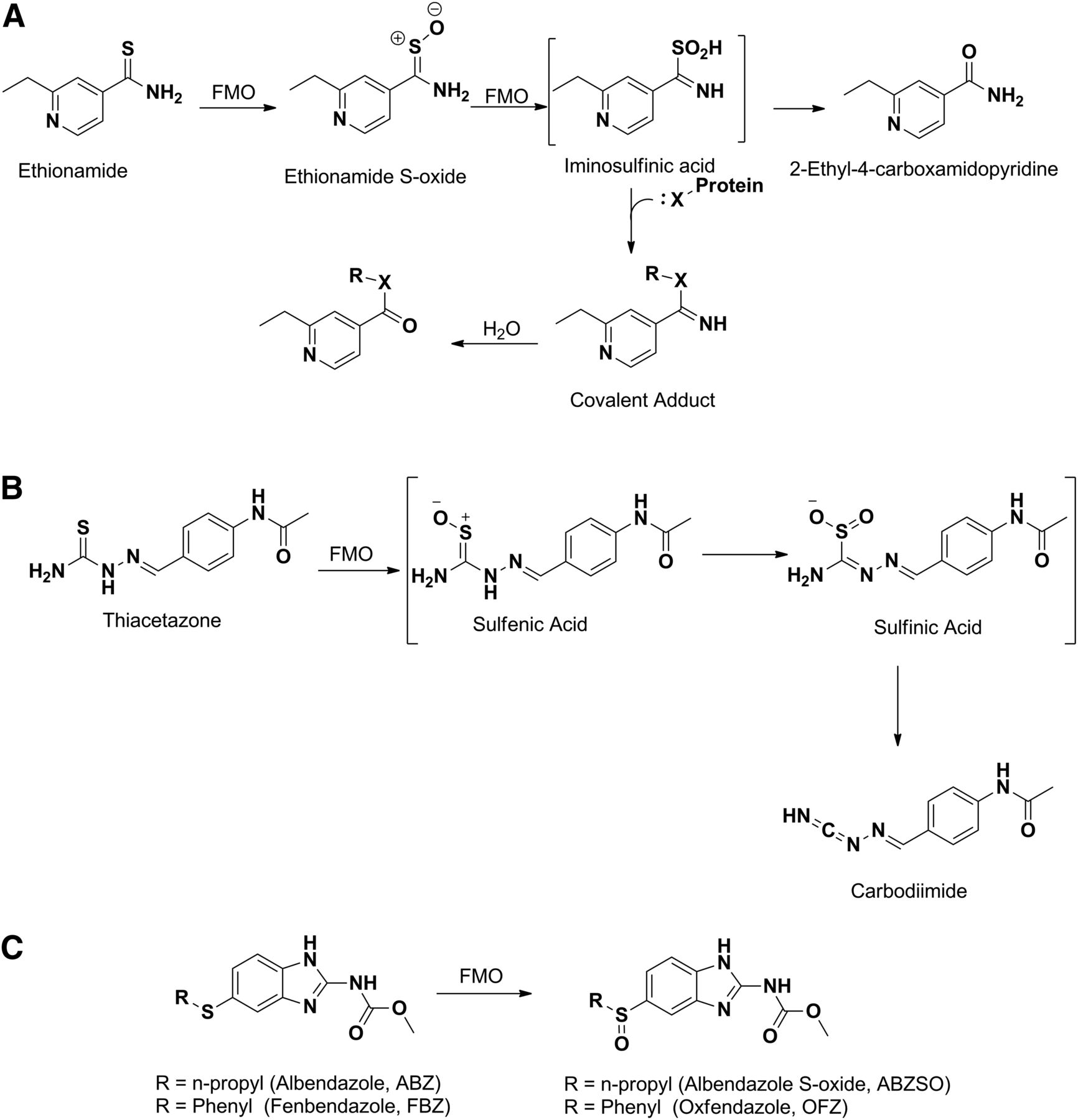
Metabolism of ethionamide (A), thiacetazone (B), and oxidation of albendazole and fenbendazole (C) to their respective S-oxide metabolites by FMO.
Aldehyde Oxidases and Xanthine Oxidases
AO and xanthine oxidase (XO) belong to the molybdenum hydrolase family and are involved in the oxidation of aldehydes and heterocycles. The effects of a concerted effort to reduce P450-mediated metabolism in xenobiotics by incorporating additional heterocycles in new chemical entities may have directly affected the role of AO in drug metabolism, because the effort indirectly resulted in an increase in the number of compounds containing an AO pharmacophore (Obach, 2004; Obach et al., 2004). The predominant site of metabolism is most often oxidation of a carbon atom that is next to the heteroatom of an aromatic ring system. A crystal structure of a mouse AO isoform (AOX3) was recently solved, with additional computational approaches being used to characterize the active site binding properties of human AO (Coelho et al., 2012, 2015). Recently, a significant amount of research has been geared toward increasing the field of knowledge around substrates and inhibitors of AOs. Traditionally, compounds such as isovanillin, hydralazine, and chlorpromazine have been used as selective inhibitors to characterize the contributions of AO to a compound’s metabolism (Obach, 2004). More recently, examples of the use of selective probe substrates of AO in hepatocytes such as carbazeran have also been reported (Hutzler et al., 2012). Examples of novel AO-catalyzed metabolic pathways also exist, as in the case of the AO-catalyzed amide hydrolysis exhibited with GDC-0834 [(R)-N-(3-(6-((4-(1,4-dimethyl-3-oxopiperazin-2-yl)phenyl)amino)-4-methyl-5-oxo-4,5-dihydropyrazin-2-yl)-2-methylphenyl)-4,5,6,7-tetrahydrobenzo[b]thiophene-2-carboxamide], an inhibitor of Bruton's tyrosine kinase (Sodhi et al., 2015).
Recent evidence has also suggested a role for AOs metabolism in species-dependent metabolic profiles, as observed with methotrexate, GDC-0834, or ripasudil, an inhibitor of Rho-associated coiled coil-containing protein kinase (Liu et al., 2011; Choughule et al., 2015; Isobe et al., 2016). In certain cases, specific arguments have been made for the use of higher preclinical species being more relevant to predict human AO metabolism, as in the case of the epidermal growth factor receptor inhibitor BIBX1382 [N8-(3-chloro-4-fluorophenyl)-N2-(1-methyl-4-piperidinyl)-pyrimido[5,4-d]pyrimidine-2,8-diamine dihydrochloride] (Fig. 4A) (Hutzler et al., 2014). Like P450, the contribution of non-P450 enzymes to drug metabolism can be significant and affect the overall development of the drug, including instances in which extensive metabolism by AO has led to clinical failures due to high clearance leading to unacceptable PK properties or due to safety-related issues. For instance, development of BIBX1382 and FK3453 [6-(2-amino-4-phenylpyrimidine-5-yl)-2-isopropylpyridazin-3(2H)-one] was discontinued because of their rapid AO-mediated elimination and resulting poor bioavailability in humans (Dittrich et al., 2002; Akabane et al., 2011). BIBX1382 (Fig. 4A) is a pyrimido-pyrimidine derivative that was selected for further development based on its preclinical profile and excellent pharmacokinetic properties in rats and mice (absolute bioavailability between 50% and 100%). However, pharmacokinetic studies in the clinic after single oral dosing showed a very poor bioavailability of approximately 5% (Dittrich et al., 2002). The poor exposure of BIBX1382 was attributed to conversion of the molecule to its inactive C, which significantly exceeded the plasma concentrations of the parent. Similarly, for FK3453 (Fig. 4A), despite favorable results in the preclinical species, the circulating concentrations of FK3453 in humans were extremely low as a result of AO-mediated oxidation of the pyrimidine moiety to the corresponding hydroxylated metabolite (Akabane et al., 2011). Other examples of AO-related clinical failures include carbazeran (a cardiac stimulant) and RO-1 (a p38 kinase inhibitor) that have been discontinued because of unexpectedly poor exposure in humans (Fig. 4B) (Kaye et al., 1984, 1985; Zhang et al., 2011).

(A) Metabolism of BIBX1382, FK3453, and idelalisib by non-P450 enzymes. (B) Structures of carbazeran and RO-1, two compounds discontinued after initiation of clinical trials as a result of poor oral exposure attributed to non-P450 enzymes.
These species-dependent metabolic profiles can also manifest themselves in terms of a drug’s safety profile. From a clinical safety standpoint, the onset of renal toxicity has been attributed to the generation of insoluble metabolites by AOs in a number of cases. For example, development of JNJ-38877605 (6-[difluoro-[6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]methyl]quinoline) was halted because of clinically observed renal toxicity, caused by the AO-catalyzed formation of insoluble metabolites in a species-dependent manner (Lolkema et al., 2015). A similar scenario was reported for SGX523 (6-[[6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]sulfanyl]quinoline), a small molecule inhibitor of Met, a tyrosine kinase inhibitor involved in tumor angiogenesis (Fig. 5) (Infante et al., 2013). Both of these c-Met inhibitors are substrates of AO and are converted to their respective quinolone metabolites via oxidation of the carbon atom adjacent to the nitrogen atom in the quinoline ring (Diamond et al., 2010; Lolkema et al., 2015). Poor solubility of the quinolone metabolite leads to its crystallization in the kidney, subsequently resulting in the observed renal toxicity in some species and humans.

AO-mediated metabolism of c-Met inhibitors JNJ38877605 and SGX-523 to their respective insoluble quinolone metabolites.
Although the role of AO in drug interactions is fairly limited to date, it is quite plausible that this could change in the near future as a result of the increasing number of xenobiotics cleared by AO. For example, zaleplon, idelalisib (Fig. 4A), and lenvatinib are all reported to have a significant portion of the metabolism dependent on AO (Zientek et al., 2010; Robeson et al., 2013; Inoue et al., 2014). Indeed, the inhibition of both the AO- and CYP3A-dependent metabolic pathways of zaleplon by cimetidine has been noted (Renwick et al., 2002). Furthermore, regularly consumed AO inhibitors such as epicatechin gallate and epigallocatechin gallate, which are found in green tea, may have the potential to cause clinically relevant drug interactions (Barr et al., 2015). Recent reports have also shown an AO-catalyzed metabolite of phosphoinositide 3-kinase δ inhibitor idelalisib to play an inhibitory role in the drug–drug interaction observed after administration of idelalisib with midazolam, which resulted in significant increases in midazolam exposures of 138%, 355%, and 437% for the Cmax, the area under the plasma concentration-time curve from time 0 to the time of the last quantifiable concentration, and the area under the plasma concentration-time curve from time 0 extrapolated to infinity, respectively (Jin et al., 2015). In vitro studies show that the oxo-metabolite is a time-dependent inhibitor of CYP3A4, with an inhibition constant of 0.2 µM and a rate of inactivation of 0.033 min−1. In addition, circulating concentrations of this metabolite are approximately 60% greater than the parent drug in humans.
On a positive note, non-P450–mediated drug oxidation or reductions can also lead to pharmacologically active metabolites that can significantly or entirely contribute to the overall therapeutic effect of a parent drug. The role of AO/XO in the conversion of drugs and or prodrugs into active metabolites has been well documented. Classic examples in this category include non-P450–catalyzed activation of antiviral guanine derivatives famciclovir (used in the treatment of herpes) and deoxyacyclovir, which are converted to their active entities penciclovir and acyclovir (Krenitsky et al., 1984; Pue et al., 1994; Clarke et al., 1995; Rashidi et al., 1997). Famciclovir is first deacetylated via rapid esterase-mediated hydrolysis to a penultimate metabolite, 6-deoxypenciclovir, which is subsequently oxidized to penciclovir (Fig. 6A). Studies by Clarke et al. (1995) and Rashidi et al. (1997) indicate that the oxidation of 6-deoxypenciclovir to penciclovir is catalyzed by AO. Similarly, the acyclic guanine analog 6-deoxyacyclovir, a widely used antiherpetic agent, is readily oxidized to the active acyclovir by XO (Fig. 6B). It is important to note that although deoxyacyclovir is also metabolized by AO, these metabolites lack activity (Krenitsky et al., 1984). Conversion of allopurinol, an XO inhibitor used in the treatment of gout and hyperuricemia, to oxypurinol is another example in which the oxidized product is active (Fig. 7) (Tamta et al., 2006). Although the metabolite is less potent than the parent (approximately 10-fold lower potency than the parent), it has a longer half-life and higher circulating concentrations compared with the parent, allowing the metabolite to contribute to the overall activity of the parent (Day et al., 2007).

Metabolism of famciclovir to penciclovir by AO (A) and metabolism of 6-deoxyacyclovir to acyclovir by XO (B).

Metabolism of allopurinol to oxypurinol by AO.
Carboxylesterases
CESs are primarily responsible for the metabolism of ester moieties in both endogenous and exogenous substrates (Lotti et al., 1983; Munger et al., 1991; Kaphalia et al., 2004). The enzymes are also capable of hydrolyzing amides and thioesters and are shown to be involved in the transesterification of multiple compounds (Boyer and Petersen, 1992; Bourland et al., 1997; Humerickhouse et al., 2000; Nishi et al., 2006). Among drug-metabolizing enzymes, CESs display one of the greater disparities in terms of activity between humans and preclinical species (Lotti et al., 1983; Takai et al., 1997). Much of the recent work on CESs has focused on the expression and regulation of the enzymes (CES1 and CES2). In terms of drug metabolism, efforts to characterize the CES-dependent pathways of compounds such as dabigatran etexilate, sacubitril, cabazitaxel, and angiotensin-converting enzyme inhibitors (e.g., ramipril and trandolapril) have been reported in recent years (Laizure et al., 2014; Thomsen et al., 2014; Tang et al., 2015; Shi et al., 2016) Furthermore, in vitro methods aimed at identifying selective CES inhibitors to use for human esterase phenotyping efforts and those to compare CES activity in immortalized cell lines from liver, intestine, and kidney sources have been undertaken (Lamego et al., 2015; Umehara et al., 2016).
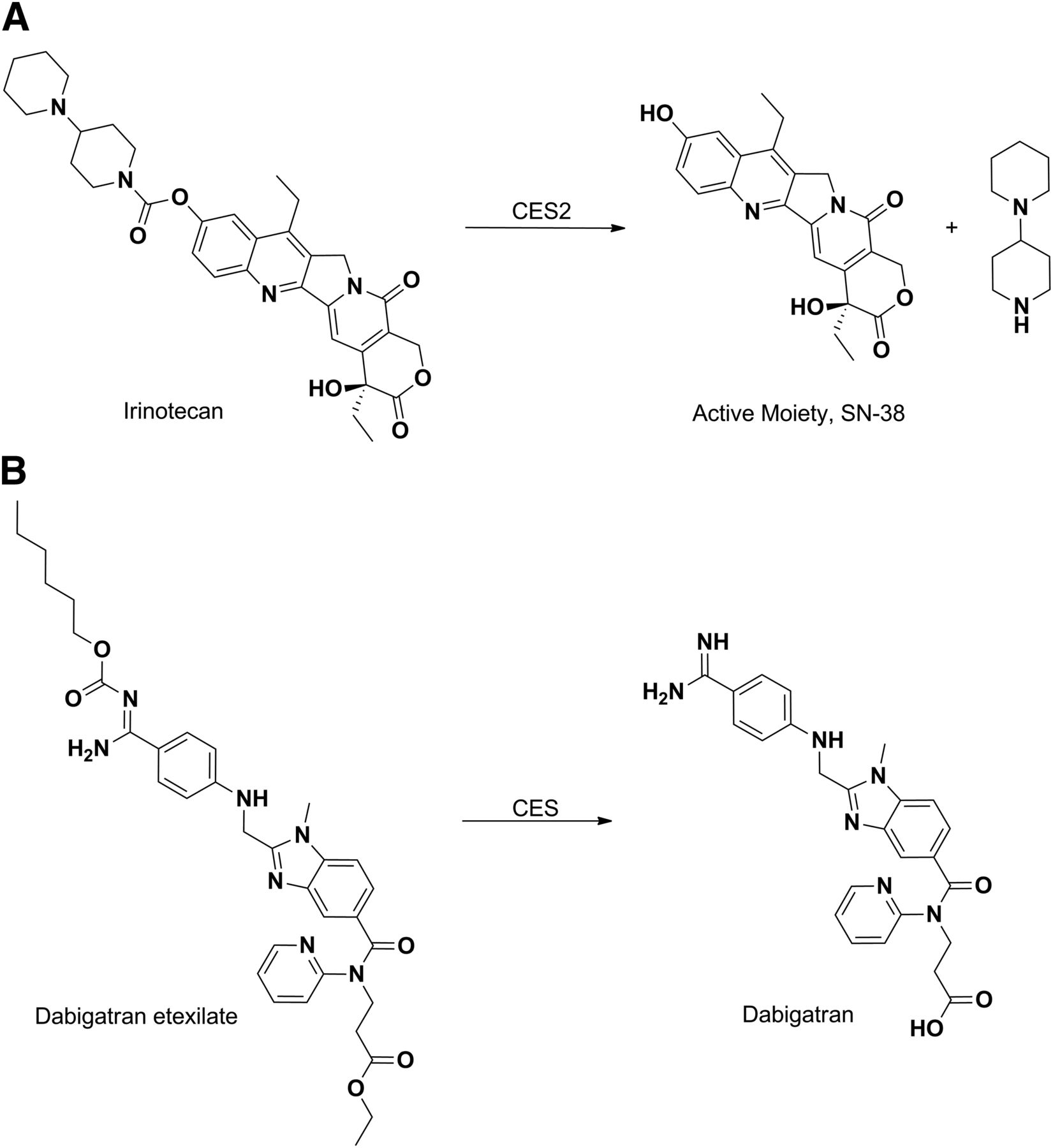
Esters and carbamates of carboxylic acid and hydroxyl functionalities are generally converted back to their respective active acids or alcohol by CESs (Beaumont et al., 2003; Ettmayer et al., 2004; Rautio et al., 2008). Thus, ester derivatives are routinely designed by medicinal chemists to enhance oral absorption of drugs or to overcome obstacles that hinder the exposure of a parent drug and therefore its efficacy. Irinotecan represents a classic example of a prodrug that is activated by CESs (Khanna et al., 2000). Irinotecan is composed of a topoisomerase I–binding moiety [SN-38 (7-ethyl-10-hydroxy-camptothecin)], coupled to a piperidino-piperidine moiety via a carbamoyl link (Fig. 8A). Hydrolysis of the carbamoyl group by carboxyesterase 2 in the human liver releases the active moiety SN-38 (Ahmed et al., 1999; Xu et al., 2002). The anticoagulant dabigatran etexilate is a direct thrombin inhibitor and is among the more recent examples of approved ester prodrugs (Stangier et al., 2007). The drug is absorbed as an ester and then the ester and carbamoyloxy functionality is hydrolyzed to afford the active moiety dagabitran (Fig. 8B) (Blech et al., 2008; Stangier, 2008).

Hydrolysis of irinotecan (A) and dabigatran etexilate (B) to their pharmacologically active forms by CES.
Prasugrel (Fig. 9A) represents an excellent example that utilizes the capability of carboxyesterase to hydrolyze esters and to overcome the obstacle of variability in the pharmacokinetics and clinical response of its predecessor clopidogrel (Fig. 9B). Both of these drugs are members of the thienopyridine class of ADP receptor inhibitors that irreversibly bind to P2Y12 receptors and prevent platelet aggregation (Bernlochner and Sibbing, 2012). Although both compounds require activation to be effective, clopidogrel is converted to its thiolactone (the precursor to its active metabolite) by CYP2C19 in the liver, whereas prasugrel is converted to its thiolactone via CES2-catalyzed hydrolysis of the acetyl ester in the intestine (Farid et al., 2010). Furthermore, approximately 85% of clopidogrel is susceptible to hydrolytic cleavage by CES1 in the liver that converts it to an inactive acid metabolite (Tang et al., 2006; Farid et al., 2010). The variability observed in the pharmacokinetics of clopidogrel is therefore attributed to the genetic polymorphism of CYP2C19 (Farid et al., 2010; Giorgi et al., 2011). The varying efficiency in the formation of the active metabolite influences the extent of inhibition of platelet aggregation by clopidogrel and results in ischemic events in at least 25% of patients despite clopidogrel treatment (Hulot et al., 2006; Giorgi et al., 2011). In contrast, the 2-oxo-metabolite of prasugrel is formed via hydrolysis of the acetyl group by carboxyesterase (CES2) in the intestine. This general non-P450–mediated pathway is less susceptible to pharmacogenetic variability and drug–drug interactions that are observed in the case of clopidogrel coadministration with proton pump inhibitors such as omeprazole (Gilard et al., 2008). Furthermore, a fraction of the active metabolite of prasugrel resulting from oxidation of its thiolactone metabolite is formed in the intestine, which leads to rapid onset of action (Farid et al., 2010).

Metabolism of prasugrel (A) and clopidogrel (B) to their pharmacologically active metabolites.
Monoamine Oxidases
The oxidative metabolism of amine functional groups to aldehydes is catalyzed by flavin-containing monoamine oxidases (MAOs) (Greenawalt and Schnaitman, 1970; Kanazawa, 1994). The primary site of metabolism for MAOs is the brain, where a significant role exists for the enzymes in the regulation of neurotransmitters (Thorpe et al., 1987). Additional sites of metabolism include the liver and placenta. Two members of the family have been characterized (MAO-A and MAO-B), with the observed substrate and inhibitor pharmacophores differing between the two enzymes (Youdim et al., 2006). Well characterized MAO-A ligands include epinephrine, 5-hydroxytrypamine, and clorgyline (Johnston, 1968). Selective MAO-B ligands include β-phenylethylamine, benzyl amine, and deprenyl (Cesura et al., 1988). Likewise, sumatriptan (Fig. 10A), a drug used in the treatment of migraines, exhibits a bioavailability of only 14% in humans, which is attributable in part to presystemic metabolism of the drug by MAO-A (Dixon et al., 1994; Scott, 1994). Given the predominance of the MAO-A–mediated pathway, the potential exists for MAO-A inhibitors such as moclobemide to inhibit sumatriptan metabolism and lead to clinically significant drug–drug interactions. Attempts have been made to mitigate the MAO-A metabolism by designing and developing the next-generation triptans that have very little contribution of MAO in their elimination (Jhee et al., 2001).
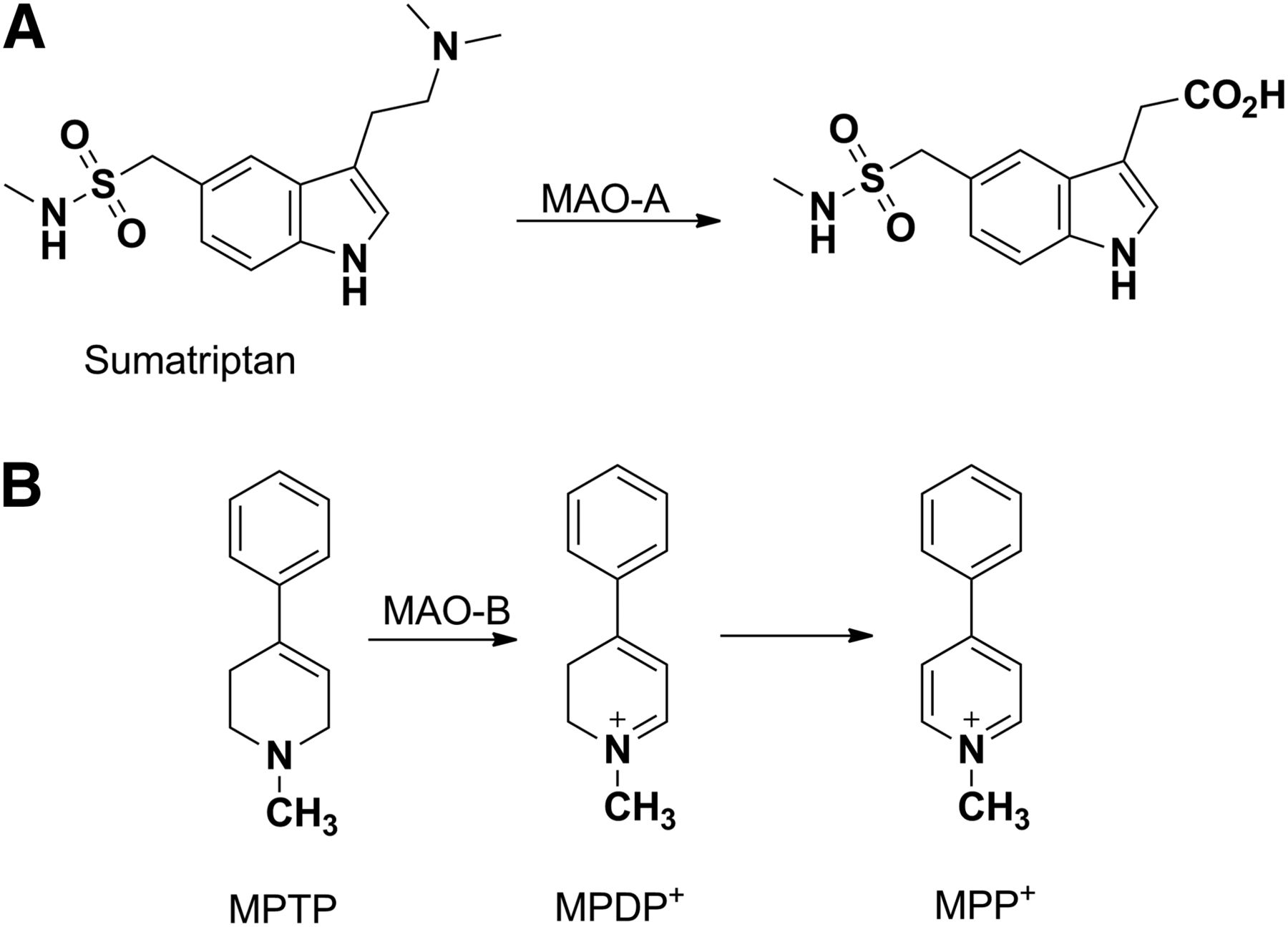
MAO-catalyzed metabolic activation of sumatriptan (A) or MPTP (B). MPDP+, 1-methyl-4-phenyl-2,3-dihydropyridinium; MPP+, 1-methyl-4-phenylpyridinium
MAO also provides an additional example of an enzyme other than P450 that has the propensity to catalyze the activation of drugs and xenobiotics to metabolites that can elicit safety concerns. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), the anti-HIV drug abacavir, and the anticonvulsant drug felbamate represent classic examples in which metabolism by non-P450 enzymes resulted in severe adverse reactions. MPTP is formed as an impurity during synthesis of an opioid analog, 1-methyl-4-phenyl-4-propionoxypiperidine, and has effects similar to those of morphine. The pathway of metabolic activation involves MAO-B–mediated oxidation of MPTP into a potent neurotoxin, 1-methyl-4-phenylpyridinium, via a 1-methyl-4-phenyl-2,3-dihydropyridinium intermediate (Fig. 10B) (Castagnoli et al., 1985; Chiba et al., 1985a). The metabolite is selectively taken up into the dopaminergic cells by a dopamine uptake transporter, where it accumulates and inhibits mitochondrial respiration and destroys dopaminergic neurons, leading to Parkinson-like symptoms (Chiba et al., 1985b; Maret et al., 1990; Gerlach et al., 1991).
Alcohol and Aldehyde Dehydrogenases
The family of ADHs encompasses a number of enzymes that metabolize primary and secondary alcohols to aldehydes and ketones, whereas ALDHs are a family of enzymes that catalyze the NADP-dependent oxidation of aldehydes to carboxylic acids (Marchitti et al., 2008). Perhaps the most well known function of the ADHs is their role in the metabolism of ethanol, with alterations in the functions of the enzymes being linked to various physiologic ramifications of alcoholism. From an endogenous standpoint, two key roles for ALDHs are the conversion of retinaldehyde to retinoic acid, a metabolic conversion that is involved in the regulation of a host of homeostatic functions, as well as the metabolism of acetaldehyde, a by-product of ethanol metabolism (Stagos et al., 2010; Singh et al., 2013). One of the key xenobiotic roles for ALDH is the bioactivation of nitroglycerin to nitric oxide, resulting in the drug’s vasodilatory effects (Chen et al., 2002; Wenzl et al., 2011).
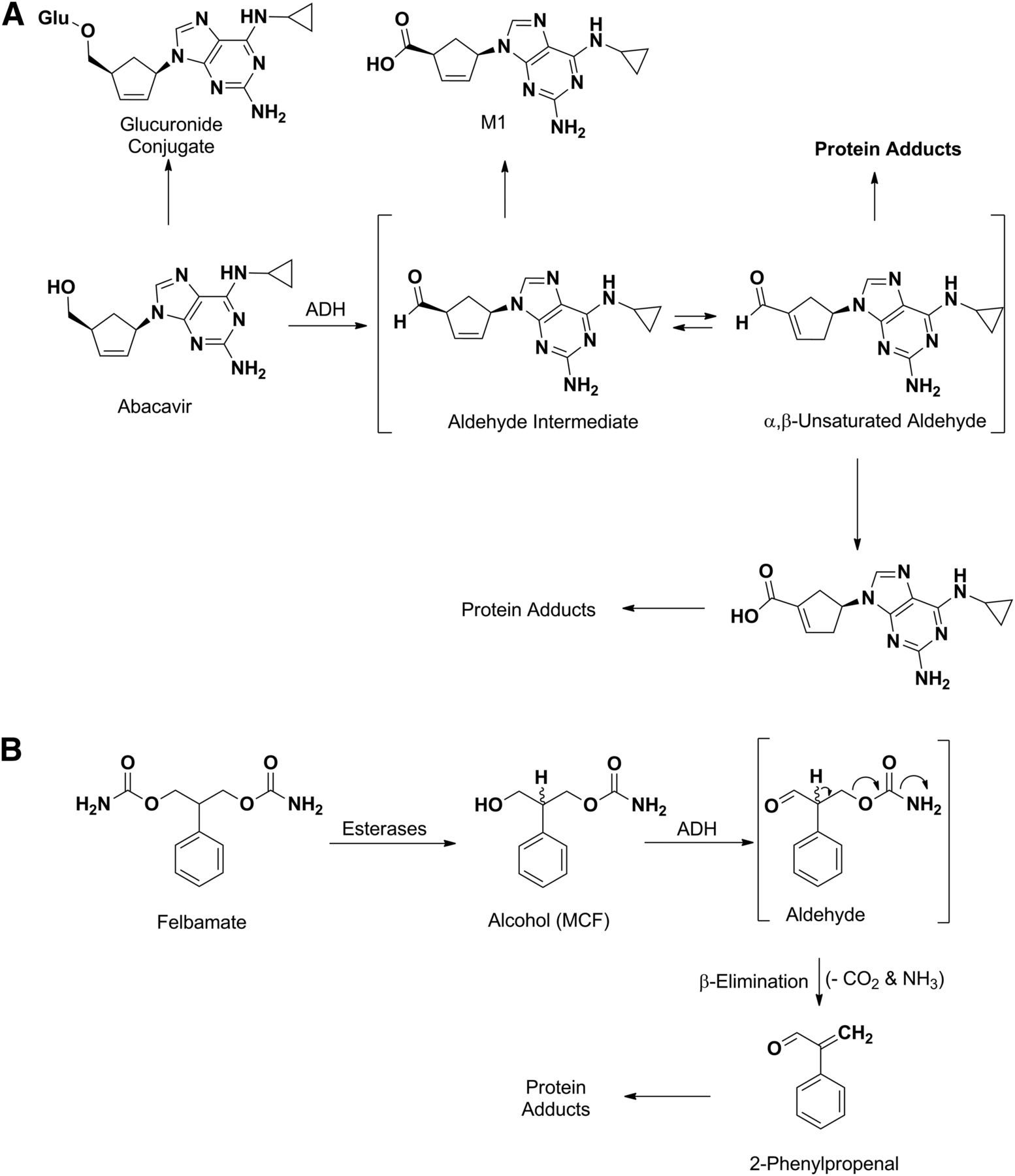
Abacavir is a nucleoside reverse transcriptase inhibitor used to prevent and treat HIV/AIDS. Although abacavir is tolerated in most patients, hypersensitivity is the main side effect in approximately 4% and is sometimes severe enough to result in death (Hetherington et al., 2001). Although the primary metabolic routes of abacavir are O-glucuronidation and formation of an unsaturated carboxylic acid, the allergic reactions caused by this drug are ascribed to the formation of α,β-unsaturated aldehyde intermediate, which is formed by ADH-catalyzed oxidation of the primary alcohol followed by isomerization of the double bond (Fig. 11A) (Walsh et al., 2002). This can react with proteins and other macromolecules in a Michael-type mechanism to form adducts that are possibly immunogenic (Grilo et al., 2013, 2014). The formation of the aldehyde intermediate was confirmed by trapping it as an oxime, through the addition of methoxylamine into the incubation mixture, whereas the role of ADH in the aldehyde formation was confirmed when the covalent adducts were blocked by the ADH inhibitor 4-methylpyrazole (Walsh et al., 2002).

ADH-mediated bioactivation of abacavir (A) and felbamate (B). MCF, monocarbamate felbamate.
Felbamate, an antiepileptic drug approved in 1993, causes aplastic anemia and possibly liver toxicity (Pellock, 1999; Pellock et al., 2006). The adverse event is attributed to the formation of a 2-phenylpropenal intermediate that can covalently bind to proteins (Dieckhaus et al., 2002; Kapetanovic et al., 2002; Roller et al., 2002). The bioactivation pathway leading to this intermediate involves several non-P450 enzymes (Fig. 11B). The first step is the conversion of felbamate to a primary alcohol (monocarbamate felbamate) intermediate that is formed by esterase- or amidase-mediated hydrolysis of the carbamate moiety. This undergoes ADH-catalyzed oxidation to the corresponding aldehyde, followed by β-elimination of the carbamoyloxy group to afford the offending moiety (2-phenylpropenal) (Dieckhaus et al., 2000; Kapetanovic et al., 2002).
Epoxide Hydrolases
Epoxide hydrolases are classified into two classes, soluble epoxide hydrolase and microsomal epoxide hydrolase, and are involved in the metabolism of epoxides to diols (Harris and Hammock, 2013; Václavíková et al., 2015). The enzymes are primarily expressed in the liver, although they can be found in many other tissues throughout the body (Pacifici et al., 1988; Coller et al., 2001). Microsomal epoxide hydrolase plays a more prominent role in drug metabolism, whereas soluble epoxide hydrolase generally regulates the concentrations of endogenous fatty acid epoxides in the cytosol (McKay et al., 1995). Furthermore, evidence of microsomal epoxide hydrolase activity at or near the blood–brain barrier has been reported (Ghersi-Egea et al., 1994). Carbamazepine, phenobarbital, and phenytoin are three xenobiotics with well characterized metabolic pathways involving epoxide hydrolase, with recent evidence suggesting that single nucleotide polymorphisms in the microsomal epoxide hydrolase gene EPHX1 influence carbamazepine exposure in vivo (El-Sherbeni and El-Kadi, 2014; Zhu et al., 2014; Daci et al., 2015).
Carbonyl Reductases/Aldo-Keto Reductases
The reduction of carbonyl moieties found in xenobiotics can be catalyzed by a number of different enzymes, with the carbonyl and aldo-keto reductases being two of the primary enzyme families involved. Of key importance is the biologic or pharmacological activity often conferred by the presence of a carbonyl moiety in a molecule; as such, the reductases can play a key role in regulating the pharmacological activity of such molecules (Oppermann, 2007). Carbonyl reductases are cytosolic enzymes that claim primarily ketone- and quinone-containing molecules as their substrates (Ris and von Wartburg, 1973). Primary sites of expression include the liver, central nervous system, and placenta (Wirth and Wermuth, 1992). Examples of carbonyl reductase substrates include menadione, doxorubicin, and daunorubicin.
As mentioned above, carbonyl reductase–catalyzed reduction of an active carbonyl group yields alcohol metabolites that may also contribute to the overall efficacy of the parent (Malátková and Wsól, 2014). Some examples are loxoprofen (Tanaka et al., 1983; Noguchi et al., 2005), befunolol (Tohno et al., 1979), and acetohexamide (Fig. 12A) (McMahon et al., 1965). Since reduction is the primary pathway for these drugs, their potency in humans is related to their respective alcohol metabolite (Ohara et al., 1995). Pentoxifylline (Fig. 12B) is another example in which reduction of the keto group in the molecule leads to a mixture of secondary alcohol metabolites (R- and S-enantiomers) (Lillibridge et al., 1996). The S-enantiomer is the major circulating metabolite and exhibits pharmacological activity that is similar to that of the parent; in contrast, the R-isomer (also called lisofylline) has completely distinct pharmacological properties (Yang et al., 2005).
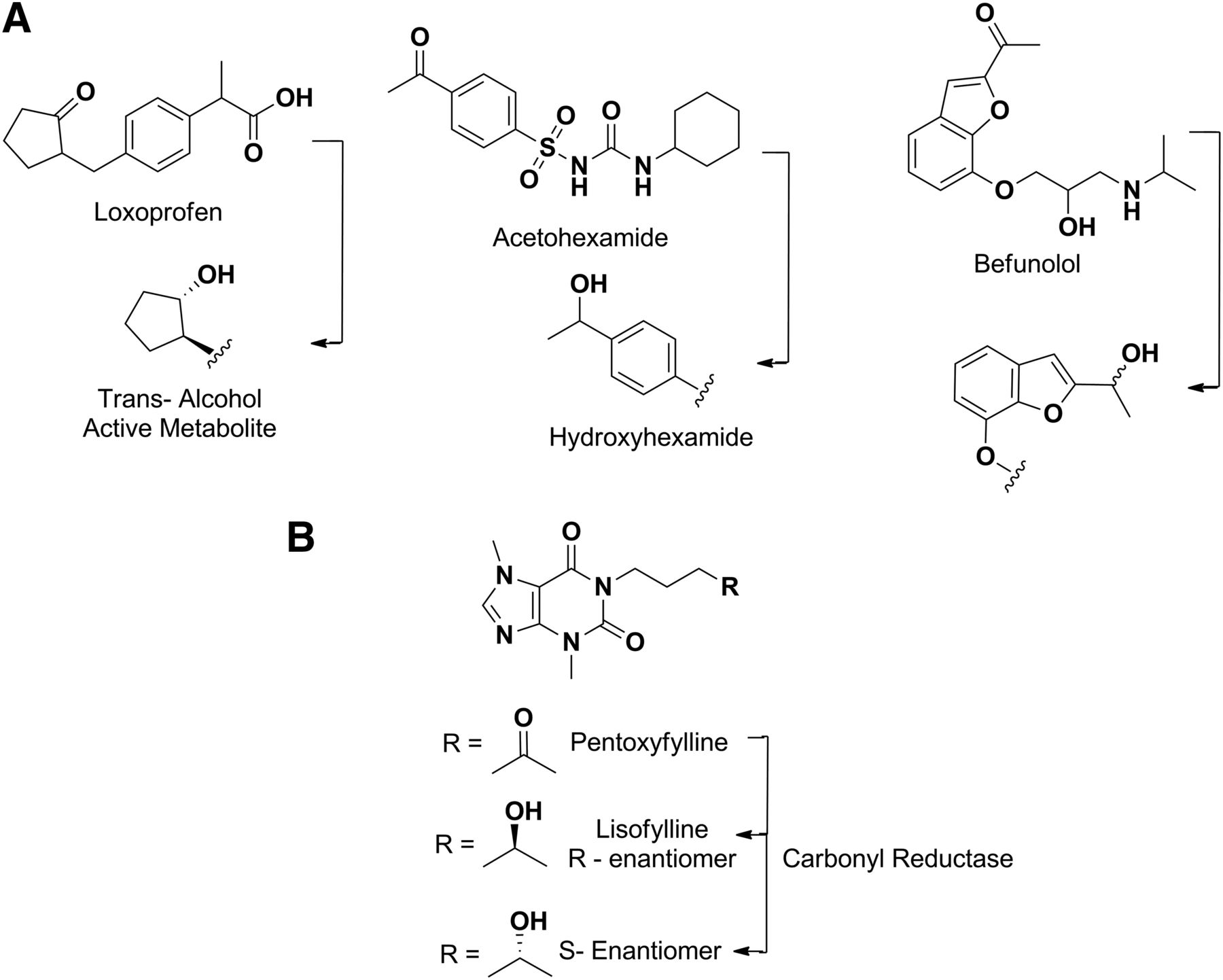
Metabolism of loxoprofen, acetohexamide and befunolol by carbonyl reductases (A). Stereoselective formation of pentoxyfylline metabolites by carbonyl reductase (B).
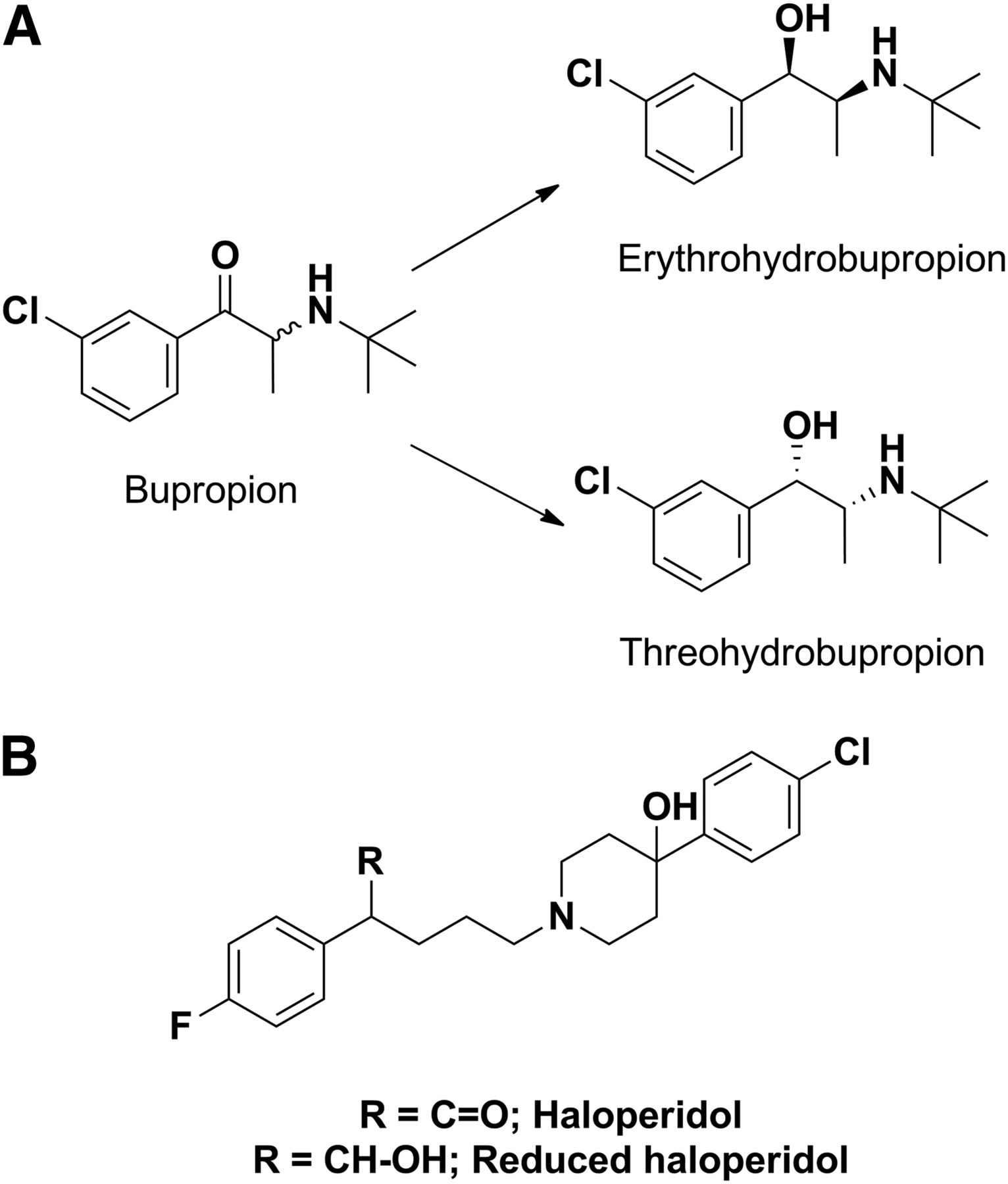
Similar to what was noted for acyl glucuronide inhibitors of CYP2C8, other non-P450s that play a role in catalyzing functionalization reactions (phase I reactions) have also produced metabolites that inhibit P450. For instance, inhibition of CYP2D6 by bupropion in the clinic and in vitro is partially attributed to threo-hydrobupropion and erythro-hydrobupropion metabolites (Fig. 13A) formed from carbonyl reductase–catalyzed reduction of the keto group (Reese et al., 2008). These reduced products have 4- and 12-fold lower inhibition constant values for CYP2D6, respectively, compared with the parent bupropion. Similarly, Shin et al. (2001) have shown that a reduced haloperidol metabolite (a carbonyl reductase mediated metabolite of haloperidol) is a more potent inhibitor of CYP2D6 compared with haloperidol (Fig. 13B).

(A) Structures of bupropion and its carbonyl reductase–catalyzed metabolites. (B) Structure of haloperidol, a substrate of aldo-keto reductase.
In an analogous fashion, the aldo-keto reductase family utilizes NADPH in the metabolism of aldehydes and ketones to the resulting alcohol derivative, which can subsequently undergo phase II metabolism to facilitate elimination. Well known substrates of aldo-keto reductases include 4-hydroxynonenal, oracin, metyrapone, haloperidol, and warfarin (Barski et al., 2008). A significant number of crystal structures for this family of enzymes have been solved, allowing for an in-depth understanding of the structural features that confer their ligand binding properties.
Conclusions
The characterization of the metabolic pathways for a given drug is a cornerstone in the discovery and development process of pharmaceutical compounds. A number of key historical and recent examples, primarily of a non-P450 nature, were presented in this commentary as a backdrop to the research included in this special section. Moving forward, an increased understanding of P450 and non-P450 enzymology, together with the development of new in vitro and in vivo tools to characterize these pathways, should allow for more complete assessments of the metabolic pathways of novel therapeutics at earlier stages of their development process. The likely outcome of such an assessment may be the mitigation of some of the late-stage drug interactions or toxicological findings that occur as a result of non-P450 metabolism. Ultimately, the expanded knowledge base should serve to provide more efficient and scientifically robust approaches to the rational design of safer and more efficacious drugs.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Foti, Dalvie.
Footnotes
- Received May 24, 2016.
- Accepted June 10, 2016.
dx.doi.org/10.1124/dmd.116.071753.
Abbreviations
- ADH
- alcohol dehydrogenase
- ALDH
- aldehyde dehydrogenase
- AO
- aldehyde oxidase
- BIBX1382
- N8-(3-chloro-4-fluorophenyl)-N2-(1-methyl-4-piperidinyl)-pyrimido[5,4-d]pyrimidine-2,8-diamine dihydrochloride
- CES
- carboxylesterase
- ER
- endoplasmic reticulum
- FK3453
- 6-(2-amino-4-phenylpyrimidine-5-yl)-2-isopropylpyridazin-3(2H)-one
- FMO
- flavin-containing monooxygenase
- GDC-0834
- (R)-N-(3-(6-((4-(1,4-dimethyl-3-oxopiperazin-2-yl)phenyl)amino)-4-methyl-5-oxo-4,5-dihydropyrazin-2-yl)-2-methylphenyl)-4,5,6,7-tetrahydrobenzo[b]thiophene-2-carboxamide
- HOIDPN
- N-hydroxy-3,3′-iminodipropionitrile
- IDPN
- iminodipropionitrile
- JNJ-38877605
- 6-[difluoro-[6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]methyl]quinoline
- MAO
- monoamine oxidase
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- P450
- cytochrome P450
- SGX523
- 6-[[6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]sulfanyl]quinoline
- SN-38
- 7-ethyl-10-hydroxy-camptothecin
- UGT
- UDP-glucuronosyltransferase
- XO
- xanthine oxidase
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Cytochrome P450 Enzymes, UDP-Glucuronosyltransferases, and Sulfotransferases
- Flavin-Containing Monooxygenases
- Aldehyde Oxidases and Xanthine Oxidases
- Carboxylesterases
- Monoamine Oxidases
- Alcohol and Aldehyde Dehydrogenases
- Epoxide Hydrolases
- Carbonyl Reductases/Aldo-Keto Reductases
- Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters