


Abstract
Generating accurate in vitro data is crucial for in vitro to in vivo extrapolation and pharmacokinetic predictions. The use of human embryonic kidney (HEK) 293 cells overexpressing organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in protein-free buffer and 100% human plasma incubations was explored for the uptake of four OATP substrates: pravastatin, rosuvastatin, repaglinide, and pitavastatin. Differences were observed for each parameter [unbound Michaelis constant (Km,u), Vmax, intrinsic clearance (CLint), and unbound passive diffusion Pdif,u] obtained from the buffer and plasma incubations in both cells, and the fold differences were greatest for the highly protein bound compounds. The fold change in Km,u values ranged from 1.91 to 619, and the fold change in Vmax values ranged from 1.22 to 97.4. As a result, in both cells, the CLint values generated in the plasma incubations were higher by 0.762- to 31.7-fold than the values generated in the protein-free buffer. The passive diffusion was also higher in the plasma incubations for all four compounds, with a fold difference range of 1.73–23.4. These shifts in the presence and absence of human plasma suggest that plasma proteins may play a role in both the active uptake and passive diffusion processes. The results also support the idea of a transporter-induced protein-binding shift, where high protein binding may not limit the uptake of compounds that have high affinity for transporters. The addition of plasma to incubations leading to higher CLint values for transporter substrates helps mitigate the underprediction commonly noted with in vitro to in vivo extrapolation.
SIGNIFICANCE STATEMENT The current investigation brings a new perspective on how to mitigate the underprediction commonly noted with in vitro to in vivo extrapolation for OATP substrates by using HEK293 cells overexpressing OATP1B1 and OATP1B3. It also supports the idea of a transporter-induced protein-binding shift, where high protein binding may not limit the uptake of compounds that have high affinity for transporters.
Introduction
Membrane transporters are known to play a key role in the absorption, distribution, and elimination of drugs as well as be determinants of their safety and efficacy profiles (Giacomini et al., 2010). Two such transporters, organic anion transporting polypeptide (OATP) 1B1 and OATP1B3, are expressed in the sinusoidal membrane of hepatocytes and mediate the uptake of both endogenous substrates as well as numerous drugs including statins (Kalliokoski and Niemi, 2009).
During the drug discovery and development process, accurate in vitro kinetic characterization of compounds is crucial for predicting specific pharmacokinetic parameters with in vitro to in vivo extrapolation (IVIVE) as well as for predicting concentration-time profiles using physiologically based pharmacokinetic (PBPK) modeling (Watanabe et al., 2009). However, limited understanding of IVIVE has brought challenges for such predictions with uptake transporter substrates. There are gaps in the field including the following: 1) how to incorporate the in vitro data generated in various systems (hepatocytes vs. cell lines); 2) how to incorporate different types of in vitro data generated [intrinsic clearance (CLint)] from substrate depletion studies (where the loss of compound from the incubation media into cells is measured) vs. Vmax/Km (where Km is the Michaelis constant) from more robust uptake experiments); 3) how to incorporate the transporter expression levels measured in various in vitro systems; and 4) how to determine if the in vitro systems used are physiologically relevant.
While traditional uptake experiments are conducted in protein-free buffer, and there is the assumption that only free drug is available for uptake, in the 1980s several single-pass liver perfusion studies found that highly bound ligands had more efficient hepatic uptake than could be accounted for by just their unbound concentrations (Forker and Luxon, 1981, 1983; Weisiger and Ma, 1987; Tsao et al., 1988). The hypotheses proposed to explain this protein-facilitated uptake phenomenon were recently reviewed (Poulin et al., 2016; Bowman and Benet, 2018; Bteich et al., 2019) and include the rate-limiting dissociation of ligand from the protein-ligand complex, rate-limiting diffusion of ligand through the unstirred water layer, interactions with the hepatocyte cell surface, and a transporter-induced protein-binding shift. In vitro investigations have also found more accurate predictions when using plasma in incubations to account for protein-facilitated uptake (Shibata et al., 2002; Blanchard et al., 2004; Mao et al., 2018). In a recent study, incorporating kinetic uptake data generated using plateable human hepatocytes with human plasma for the OATP substrate pravastatin enabled a PBPK model to simulate the intravenous and oral pharmacokinetic profiles of the drug successfully without incorporating a scaling factor (Mao et al., 2018). This demonstrated a successful approach for translating in vitro OATP uptake transporter data to in vivo, with the hope of utilizing future in vitro data for accurate human pharmacokinetic predictions.
In the continuing effort to understand the translation of in vitro data to in vivo and the plasma effect with uptake transporters, the use of human embryonic kidney (HEK) 293 cells transfected with OATP1B1 and OATP1B3 was explored in the current investigation. These cells were chosen for two reasons. 1) While HEK293 cells are typical cell lines used in the drug discovery stage, serving as an economical option to qualitatively address if a new chemical entity is a substrate for OATPs, there is an IVIVE gap for quantitative translation. 2) Given that any protein-facilitated uptake noted with HEK293 cells would not be due to interactions with the hepatocyte cell surface, the system serves as a tool to assess the various aforementioned hypotheses. The objective of this study was to evaluate the uptake of known OATP substrates, with both low and high protein binding, in HEK293 cells transfected with OATP1B1 and OATP1B3 in buffer and human plasma incubations.
Materials and Methods
Materials.
Pitavastatin calcium and repaglinide were purchased from Abcam (Cambridge, UK), pravastatin sodium was purchased from TCI America (Portland, OR), and rosuvastatin was purchased from BioVision Inc. (Milpitas, CA). Propranolol and probenecid were purchased from Sigma Chemical Co (St. Louis, MO). Corning TransportoCells Cryopreserved SLC Transporter Cells [human OATP1B1*1a (lot 6034125), OATP1B3 (lot 5278015), and control cells (lot 6075312)] were purchased from Corning (Corning, NY) and used for uptake studies. Human Plasma Medium-A (100% human plasma) and hepatocyte rinse medium were purchased from In Vitro ADMET Laboratories, Inc. (Columbia, MD). Poly-D-lysine–coated 96-well plates were purchased from Greiner Bio-One (Monroe, NC). Fetal bovine serum was purchased from VWR (Radnor, PA), and all other cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA). A single-use rapid equilibrium dialysis plate with inserts was purchased from Thermo Fisher Scientific.
Uptake in OATP1B1- and OATP1B3-Overexpressing Cells.
Uptake studies were performed in triplicate following the TransportoCells manual. Briefly, after thawing, cells were seeded onto poly-D-lysine–coated 96-well plates with a seeding density of 100,000 cells per well in plating media consisting of Dulbecco’s modified Eagle’s medium (high glucose) with minimal essential medium nonessential amino acid solution (100X) and FBS. After incubation in humidified atmosphere with 5% CO2 at 37°C for 3 hours, the cells were re-fed with plating media supplemented with 2 mM sodium butyrate. Uptake experiments were initiated 24 hours after plating.
To begin uptake experiments, plating media were removed from the cells, and cells were washed once with either Hanks’ balanced salt solution buffer (with 10 mM HEPES, pH 7.4) or human plasma. Drug solutions of either protein-free Hanks’ balanced salt solution buffer or human plasma were added for 1 minute. The incubation time of 1 minute was selected based on time course experiment results for both medium conditions (results not shown). For studies in buffer, uptake was terminated by removing the dosing solution and washing cells twice with ice-cold PBS. For studies in plasma, uptake was terminated similarly, but by washing three times with ice-cold hepatocyte rinse medium. The following concentrations were tested in both cells with buffer and plasma: 200, 100, 50, 25, 12.5, 6.25, and 3.13 μM for pravastatin; 1.25, 0.625, 0.3125, 0.156, 0.0781, and 0.0391 μM for repaglinide; and 50, 25, 12.5, 6.25, 3.13, 1.56, and 0.781 μM for rosuvastatin. For pitavastatin, 10, 5, 2.5, 1.25, 0.625, 0.313, 0.156, and 0.0781 μM concentrations were tested under both conditions; however, biphasic kinetics were seen after 2.5 μM in plasma, thus the calculations were based on 2.5, 1.25, 0.625, 0.313, 0.156, and 0.0781 μM concentrations for the plasma incubations. The ranges chosen were based on previously reported values to ensure the Km value would be captured.
The viability of the cells in Hanks’ balanced salt solution buffer and human plasma was determined with a Nexcelom GigaCyte Cellometer Hepatometer. The amount of protein in each well was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA).
To measure the analyte concentrations, incubation samples were mixed with acetonitrile:water (80:20) containing internal standard (propranolol for positive ion mode and probenecid for negative ion mode), sonicated, and then centrifuged. The supernatant was mixed with 50% water and bioanalytical assays were developed to obtain analyte concentrations.
Plasma Protein Binding.
Plasma protein binding experiments were performed at two concentrations per compound (one high concentration and one low concentration based on the concentrations used in the cellular uptake experiments) with three replicates in a shaking incubator at 37°C with 5% CO2 at 450 rpm (VWR Symphony). Experiments were conducted using a single-use rapid equilibrium dialysis plate with inserts and the following set-up: 300 μl of plasma-drug mixture was added to the donor side and 500 μl buffer (PBS containing 133 mM KPi and 150 mM NaCl) was added to the receiver side. Experiments were conducted using 100% Human Plasma Medium-A (100% human plasma). Following 24-hour incubation, aliquots of samples were taken out of the rapid equilibrium dialysis device and equalized with an equal amount of either plasma or PBS (buffer was added to the donor side and plasma was added to the receiver side). After equalization, samples were quenched in sample:acetonitrile (1:3 ratio) with ice-cold high-performance liquid chromatography grade acetonitrile (EMD Millipore, Billerica, MA) and propranolol or probenecid as the positive and negative internal standards, respectively, and shaken for 15 minutes at 500 rpm (Thermo Scientific Compact Digital MicroPlate Shaker) followed by centrifugation at 3750 rpm for 15 minutes (Beckman Coulter Allegra X-12R). Subsequently, supernatants were further diluted 1:1 with high-performance liquid chromatography grade water (J.T. Baker, Center Valley, PA). The fraction unbound in plasma (fup) was calculated using the following equation: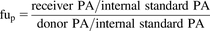 where PA is the peak area.
where PA is the peak area.
Bioanalyical Method.
All samples were analyzed with a Shimadzu Nexera X2 (LC-30AD) (Kyoto, Japan) coupled to a Sciex 6500 QTRAP mass spectrometer (Foster City, CA). Pitavastatin, repaglinide, and rosuvastatin were analyzed in positive ion mode with propranolol (260.0/116.1) as the internal standard, and pravastatin was analyzed in negative ion mode with probenecid (284.0/139.0) as the internal standard. The liquid chromatography separations were performed using a Kinetex C18 column (2.6 μm, 30 × 2.1 mm) (Phenomenex, Torrance, CA). The mobile phases consisted of water with 0.1% formic acid (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B). Additional details about the methods used can be found in Table 1.
Liquid chromatography–tandem mass spectrometry methods for pravastatin, rosuvastatin, repaglinide, and pitavastatin
Modeling of the Active Hepatic Uptake Transporter Kinetic Parameters and Passive Diffusion.
Data analysis was done using GraphPad Prism version 7 (GraphPad Software, La Jolla, CA). Unbound concentrations were used for the plots. Uptake curves from the control cells, representing passive diffusion only, were fit linearly to determine the passive diffusion corrected for protein binding (Pdif,u = Pdif/fup, where Pdif,u is the unbound passive diffusion and Pdif is the passive diffusion). The difference between uptake in the transfected cells (active + passive uptake) and uptake in the control cells (passive uptake) was plotted, representing the active uptake alone, where this curve was used to obtain the unbound Michaelis constant [(Km,u), Km,u = Km × fup] and Vmax values, assuming Michaelis-Menten kinetics. For the protein-free buffer incubations the fup value was assumed to be 1 and for the plasma incubations, the fup values were measured as previously detailed.
Results
First, the morphology and viability of the cells were tested in 100% human plasma versus protein-free buffer. While cellular clumping visually appeared to occur during the plasma incubations, the viability of both OATP1B1 and 1B3 cells was high and comparable between the two incubations (Table 2).
The viability of the OATP1B1 and OATP1B3 cells in protein-free buffer and 100% human plasma
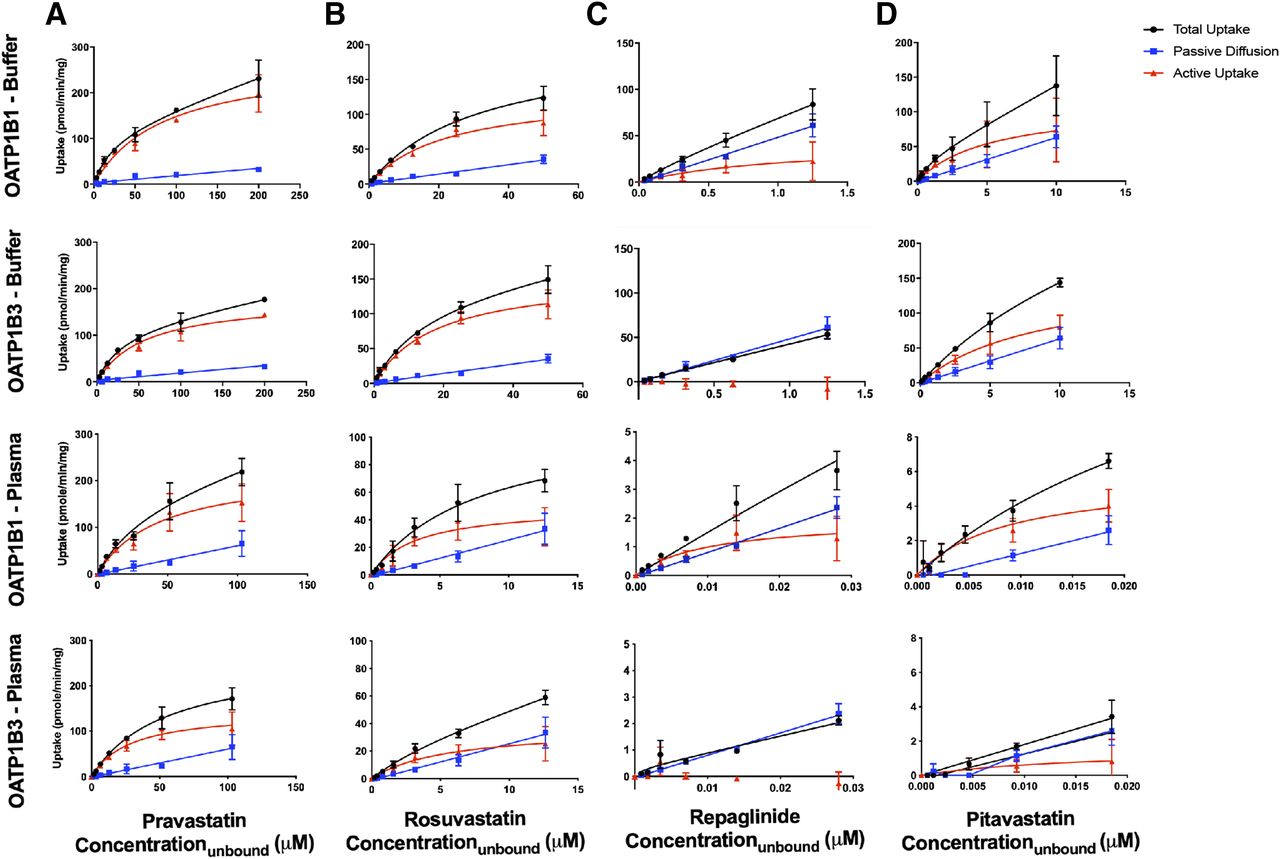
The uptake curves for four known OATP substrates under both incubation conditions are shown in Fig. 1. Active uptake was observed for pitavastatin, pravastatin, and rosuvastatin in both OATP1B1 and OATP1B3 cells in the buffer and plasma incubations. For repaglinide, active uptake was observed in the OATP1B1 cells, and the uptake in the OATP1B3 cells overlapped with the uptake in control cells, demonstrating that the drug is not an OATP1B3 substrate in the concentration range tested.

The rate of the uptake vs. the unbound concentration of pravastatin (A), rosuvastatin (B), repaglinide (C), and pitavastatin (D) in OATP1B1- and OATP1B3-overexpressing cells in buffer and human plasma. Total uptake (overexpressed cells) is depicted by circles, passive diffusion (control cells) is depicted by squares, and active uptake (the difference) is depicted by triangles.
Plasma protein binding values are reported in Table 3, and the values were in agreement with those previously reported in the literature (Pan et al., 1990; Lennernäs and Fager, 1997; Fujino et al., 1999; Hatorp et al., 2000; Plum et al., 2000; Martin et al., 2003; Watanabe et al., 2009; Jones et al., 2012). No difference in values was noted between the low and high concentrations for each drug. The fup value at the low concentration was used for subsequent data analysis.
Human plasma protein binding values of the pravastatin, rosuvastatin, repaglinide, and pitavastatin (n = 3)
The calculated Km,u, Vmax, and Pdif,u values are shown in Table 4. The Km,u values generated in the buffer aligned well with reported values in the literature. When examining the difference in Km,u values generated in buffer versus plasma incubations, there were large differences for the highly protein bound compounds. For instance, examining the OATP1B1 cell data, pravastatin with the lowest protein binding (fup = 0.516) had a Km,u fold difference of 1.91 between the two incubations (84.9 vs. 44.5 μM) and rosuvastatin (fup = 0.253) had a Km,u fold difference of 6.83 (22.2 vs. 3.25 μM), while repaglinide (fup = 0.0224) had a Km,u fold difference of 107 (1.07 vs. 0.0100 μM) and pitavastatin (fup = 0.00740) had a Km,u fold difference of 619 (5.29 vs. 0.00855 μM). For the OATP1B3 cell data, a similar trend was observed. The fold differences of Km,u values were 2.02, 3.21, and 561 for the pravastatin, rosuvastatin, and pitavastatin, respectively. The Vmax value also decreased for the highly bound compounds. Examining OATP1B1, the difference was 1.22-fold for pravastatin and 2.64-fold for rosuvastatin, compared with 21.7- and 19.5-fold for repaglinide and pitavastatin, respectively. Similar trends were seen with the OATP1B3 cells with fold differences of 1.23, 4.20, and 97.4 for pravastatin, rosuvastatin, and pitavastatin, respectively.
The Km,u, Vmax, Pdif,u, CLint, and percentage of active uptake values generated for pravastatin, rosuvastatin, repaglinide, and pitavastatin in OATP1B1 and OATP1B3 overexpressing cells in buffer and human plasma (n = 3)
CLint = Vmax/Km,u.
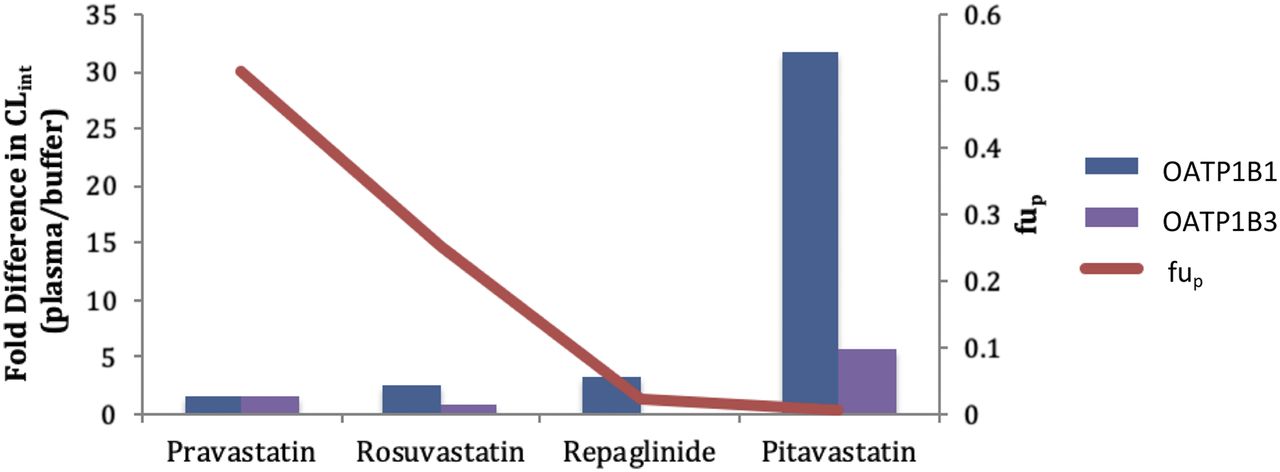
The CLint (Vmax/Km,u) value was determined for each compound and the fold differences between the values in plasma and buffer were calculated (Fig. 2). Again, the difference in CLint became greater as the free fraction decreased. For the lowest binding pravastatin, the differences in CLint values were 1.56-fold in OATP1B1 cells and 1.63-fold in OATP1B3 cells, while for the highest binding pitavastatin, the fold differences were 31.7 and 5.75, respectively. The percentage of active uptake [CLint/(CLint + Pdif,u)] was examined between the incubations as well (Table 4). The values were similar for pravastatin, rosuvastatin, and pitavastatin between the two incubation conditions for OATP1B1 cells. Repaglinide, the compound with the highest passive diffusion, had the largest increase in percentage of active uptake with the use of plasma (45.2 vs. 70.2%) in the OATP1B1 cells.

The plasma protein binding vs. the fold difference in the CLint value of pravastatin, rosuvastatin, repaglinide, and pitavastatin generated in buffer and human plasma for OATP1B1 and OATP1B3.
Discussion
Obtaining accurate in vitro data is crucial for predicting specific pharmacokinetic parameters such as clearance with IVIVE, as well as for generating reliable concentration profiles with PBPK modeling. There is often a disconnect with current IVIVE methods particularly for compounds that are substrates of transporters (Soars et al., 2007). In vivo clearance for seven OATP substrates was previously underpredicted using human hepatocytes in protein-free incubations, and predictions were poorest for those substrates that were highly protein bound (Jones et al., 2012).
Here, we evaluated HEK293 cells overexpressing OATP1B1 and OATP1B3 in protein-free buffer and 100% plasma incubations as an alternative system. Research groups have recently begun adding protein to incubations with overexpressing cell lines using, for instance, 2% human serum albumin (Fukuchi et al., 2017) or 10% fetal bovine serum (Oh et al., 2018). In this investigation, using 100% human plasma with HEK293 cells was also demonstrated to be a reasonable option since the cells maintained high viability throughout the study.
Four known substrates of OATP with a range of protein binding values were chosen to evaluate. Pravastatin, pitavastatin, and rosuvastatin were all substrates of OATP1B1 and OATP1B3, agreeing with previously reported in vitro studies (Table 4) and in vivo studies (Choi et al., 2008; Deng et al., 2008; Alam et al., 2018). Repaglinide was found to be a substrate of OATP1B1 in the concentration range tested, agreeing with previous in vitro and in vivo studies that have reported OATP1B1 to be a major determinant of its pharmacokinetics (Niemi et al., 2005; Kalliokoski et al., 2008). While an in vitro study found repaglinide to be an inhibitor of OATP1B3 (Bachmakov et al., 2008), it was not demonstrated to be a substrate here. However, a recent study found repaglinide to be an OATP1B3 substrate using longer incubation times (Fujino et al., 2018).
Differences were observed with each parameter (Km,u, Vmax, CLint, and Pdif,u) obtained from the buffer and human plasma incubations in both cells for the highly protein bound compounds. Similar to what was previously noted with the use of plasma in hepatocyte incubations (Bowman et al., 2019), the Km,u values decreased in the plasma as protein binding increased (with fold changes ranging from 1.91 to 619). The Vmax value also decreased in the plasma for the higher binding compounds (the fold changes ranged from 1.22 to 97.4). The decreases in both Km,u and Vmax, and the more dramatic differences in Km,u values led to higher CLint (Vmax/Km,u) values in the human plasma incubations for the highly bound compounds. The fold difference (plasma/buffer) was largest for the highest protein bound pitavastatin (31.6 and 5.75 for OATP1B1 and OATP1B3, respectively) compared with the differences with the other three compounds (1.56–4.94 and 0.762–1.63 for OATP1B1 and OATP1B3, respectively) (Fig. 2). This suggests that plasma proteins may play a role in the active uptake process.
The passive diffusion was also consistently higher in the human plasma incubations for all four compounds. The fold difference again was higher for pitavastatin (23.4) compared with the other three compounds (1.73–3.90), suggesting that plasma may influence the passive diffusion process as well. This observation was also previously reported in hepatocytes (Bowman et al., 2019); however, the mechanism remains unknown. In a previous study, Neuhoff et al. (2006) found that the passive apparent permeability for nontransporter substrates was comparable in the presence and absence of 4% bovine serum albumin in Caco-2 cells after taking into account the free fraction. While this is a different in vitro system with different compounds, it suggests that perhaps a component of plasma besides albumin could be leading to the increase in passive diffusion seen here. Since there was an increase in CLint, in addition to Pdif,u, when examining the percentage of active uptake shifts for the three statin compounds, generally the passive diffusion differences did not have an effect. For compounds with high plasma protein binding that are not substrates for active uptake (but are, for example, predominately metabolized), the results from protein-free incubations may be underestimated.
These Km,u/Vmax/CLint values shifted in the same way as those for a highly protein bound new chemical entity in HEK293-overexpressing cells with human serum albumin (Fukuchi et al., 2017), where Km,u decreased, Vmax decreased, and CLint increased with increasing concentrations of human serum albumin. In particular, for pravastatin the Km,u values measured in the OATP1B1- and OATP1B3-overexpressed cell lines in plasma were 44.5 and 28.5 μM, respectively, which were quite comparable to the reported Km,u value of 37 μM generated with plated human hepatocytes in plasma (Mao et al., 2018). Many research groups have also seen decreased Km,u and higher CLint values when using plasma in hepatocyte incubations (Blanchard et al., 2004, 2006). An early idea of including plasma was to eliminate the uncertainty in prediction introduced with separately measuring plasma protein binding, which improved clearance predictions (Shibata et al., 2000, 2002). Having plasma or proteins present also can account for potential protein-facilitated uptake (Miyauchi et al., 2018; Kim et al., 2019).
According to the free drug hypothesis, hepatic uptake of drug is solely dependent on the unbound concentration available at the hepatocyte surface; however, several studies have unexpectedly found uptake to be greater than the unbound concentration (Poulin et al., 2016; Bowman and Benet, 2018). While the presence of a specific albumin receptor on the hepatocyte cell surface was originally proposed, numerous studies and negative results from affinity chromatography have deemed the hypothesis less likely (Stremmel et al., 1983). Instead, the idea of more general interactions of the albumin-drug complex with the hepatocyte cell surface is frequently discussed (Poulin and Haddad, 2018).
For instance, binding of the albumin-drug complex to the cell surface may lead to a conformational change in albumin, reducing its binding affinity for the drug (Tsao et al., 1988), or ionic interactions between the protein-drug complex and the hepatocyte plasma membrane may decrease the diffusional distance for unbound ligand (Burczynski et al., 1997). However, these hypotheses focus on hepatocytes specifically and protein-facilitated uptake has been shown to occur in other cell types including myocytes (Hütter et al., 1984), adipocytes (Sorrentino et al., 1989), and in this study in HEK293 cells overexpressing transporters.
These data support a recent hypothesis called the transporter-induced protein-binding shift (TIPBS) (Bowman and Benet, 2018; Bowman et al., 2019). The idea is that if a highly protein bound drug has a higher affinity for a transporter than for a plasma protein (or the suggested hepatocyte cell surface), the transporter may be able to strip the drug from the protein before the drug dissociates itself and is at binding equilibrium. In these cases, protein binding would not be restricting the access of the compounds. While the alternative hypotheses may have been able to explain the trends seen in hepatocytes in the past, the TIPBS explanation agrees with data from hepatocytes as well as from HEK293 cells, where overexpressed transporters (not a hepatocyte cell surface) would be driving the Km,u and CLint shifts seen. To explain the Vmax decreases seen both here and by Fukuchi et al. (2017), there could still be a weaker interaction of the protein-drug complexes with the HEK293 cells leading to more complexes near the cell surface and decreasing the maximum velocity of substrate uptake, which was hypothesized with TIPBS and hepatocytes (Bowman et al., 2019).
Interesting results were found for repaglinide. Although it has the second highest protein binding of the four compounds evaluated (fup = 0.0224), compared with rosuvastatin with lower binding (fup = 0.253), repaglinide had a similar CLint shift. One simple explanation could be that the other compounds examined were statins and they can be more directly compared with each other than to repaglinide. In addition, an explanation related to a TIPBS could also explain the difference. Repaglinide had the largest percentage of passive diffusion of the four compounds in the overexpressed OATP1B1 cell line (Table 4). Given the lesser involvement of transporters and active uptake, a transporter-induced protein-binding shift would also be expected to be less. The addition of plasma did increase the percentage of active uptake for repaglinide, but it was still less than that of the other three compounds.
In conclusion, this work found changes in the kinetic uptake of OATP1B1 and OATP1B3 substrates when using HEK293 cells overexpressing transporters in plasma incubations. In general, the CLint values generated in the plasma incubations were higher than the values generated in protein-free buffer in both cells, addressing the underprediction related to OATP substrates reported previously. The results also support the recent hypothesis of a transporter-induced protein-binding shift for compounds with high protein binding and high affinity for transporters. PBPK modeling work is ongoing to provide systematic understanding of the in vitro to in vivo translation of data from overexpressed cell lines in the presence and absence of human plasma.
Acknowledgments
The authors thank Jonathan Cheong, Emile Plise, and Shu Zhang for help in the laboratory; Matthew Harwood, Iain Gardner, and Sibylle Neuhoff from Certara for discussions of plasma protein binding and the Caco-2 cell investigation; and Zhengyin Yan and Laurent Salphati for reviewing the manuscript and helpful suggestions.
Authorship Contributions
Participated in research design: Bowman, Y.-C. Chen, Y. Chen, Mao.
Conducted experiments: Bowman, E. Chen, L. Chen, Y.-C. Chen.
Performed data analysis: Bowman, L. Chen, Y.-C. Chen, Mao.
Wrote or contributed to the writing of the manuscript: Bowman, E. Chen, L. Chen, Y.-C. Chen, Liang, Wright, Y. Chen, Mao.
Footnotes
- Received August 1, 2019.
- Accepted October 28, 2019.
Abbreviations
- CLint
- intrinsic clearance
- fup
- fraction unbound in plasma
- HEK
- human embryonic kidney
- IVIVE
- in vitro to in vivo extrapolation
- Km
- Michaelis constant
- Km,u
- unbound Michaelis constant
- OATP
- organic anion transporting polypeptide
- PBPK
- physiologically based pharmacokinetic
- Pdif,u
- unbound passive diffusion
- TIPBS
- transporter-induced protein-binding shift
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}