


Abstract
The transporter-mediated uptake of drugs from blood into hepatocytes is a prerequisite for intrahepatic drug action or intracellular drug metabolism before excretion. Therefore, uptake transporters, e.g., members of the organic anion transporting polypeptide (OATP) family are important determinants of drug pharmacokinetics. Highly and almost exclusively expressed in hepatocytes are the OATP family members OATP1B1 (SLCO1B1) and OATP1B3 (SLCO1B3). Drug substrates of OATP1B1 and OATP1B3 include antibiotics and HMG-CoA reductase inhibitors (statins). It has been demonstrated that administration of two or more drugs that are substrates for these hepatic uptake transporters may lead to transporter-mediated drug-drug interactions, resulting in altered transport kinetics for drug substrates. In this study we investigated whether non-steroidal anti-inflammatory drugs (NSAIDs) and paracetamol interact with OATP1B1 and OATP1B3 using the standard substrate BSP and the drug substrate pravastatin. Using human embryonic kidney cells stably expressing OATP1B1 or OATP1B3, we demonstrated that bromosulfophthalein uptake was inhibited by diclofenac, ibuprofen. and lumiracoxib. Of interest, pravastatin uptake was stimulated by these NSAIDs, and for ibuprofen we determined activation constants (EC50 values) of 64.0 and 93.1 μM for OATP1B1- and OATP1B3-mediated uptake, respectively. Furthermore, we investigated whether NSAIDs were also substrates for OATP1B1 and OATP1B3 and demonstrated that only diclofenac was significantly transported by OATP1B3, whereas all other NSAIDs investigated were not substrates for these uptake transporters. These results demonstrated that drugs may interact with transport proteins by allosteric mechanisms without being substrates and, therefore, not only uptake inhibition but also allosteric-induced modulation of transport function may be an important mechanism in transporter-mediated drug-drug interactions.
Introduction
Uptake transporters in the basolateral membrane of human hepatocytes are important determinants for the hepatobiliary elimination of drugs (König et al., 2006; Koepsell et al., 2007; König, 2010). Furthermore, the uptake of drugs from blood into hepatocytes is a prerequisite for subsequent intracellular drug metabolism. Several members of various uptake transporter families are expressed in human liver and are located in the basolateral hepatocyte membrane (Koepsell et al., 2007; Kindla et al., 2009; Fahrmayr et al., 2010). Most uptake transporters belong to the superfamily of solute carriers (SLC superfamily) (Hediger et al., 2004). One important SLC family with members expressed in human hepatocytes is the OATP (SLCO/SLC21) family. In human liver, three OATP family members are highly expressed, namely, OATP1B1 (gene symbol SLCO1B1) (Abe et al., 1999; Hsiang et al., 1999; König et al., 2000b), OATP1B3 (SLCO1B3) (König et al., 2000a), and OATP2B1 (SLCO2B1) (Kullak-Ublick et al., 2001). Whereas OATP2B1 is expressed in other tissues also (Grube et al., 2006), OATP1B1 and OATP1B3 are predominantly, if not exclusively, expressed in hepatocytes (Hsiang et al., 1999; König et al., 2000a,b). Substrates for OATP1B1 and OATP1B3 include endogenous compounds such as bile salts and steroid hormones as well as several widely prescribed drugs such as antibiotics, cardiac glycosides, and HMG-CoA reductase inhibitors (statins) (Hsiang et al., 1999; Kullak-Ublick et al., 2001; König et al., 2006).
Because of this substrate spectrum, OATP1B1 and OATP1B3 are involved in transporter-mediated drug-drug interactions. Two simultaneously administered drugs, which are both substrates for one of these uptake transporters, may influence the uptake kinetics of one or both drugs, resulting in a change in drug plasma concentrations due to altered hepatic uptake. This result has been demonstrated in vivo (Jacobson, 2004) and studied extensively in vitro using cell systems recombinantly overexpressing OATP1B1 or OATP1B3. With use of pravastatin as the drug substrate for OATP1B1 or OATP1B3 and macrolides (Seithel et al., 2007) or oral antidiabetic drugs (Bachmakov et al., 2008), it has been demonstrated that simultaneous application of these drug combinations resulted in altered uptake kinetics for pravastatin.
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used because of their analgesic, antipyretic, and anti-inflammatory therapeutic effects. NSAIDs are acids and are mostly metabolized in hepatocytes by oxidation and conjugation to inactive metabolites and subsequently excreted into urine or into bile (Needs and Brooks, 1985; Brater, 1988). To date, knowledge about the uptake of NSAIDs from blood into hepatocytes is limited. It has been demonstrated in rats that the uptake transporter Oat2 (Slc22a7) is involved in the hepatic uptake of ketoprofen (Morita et al., 2005), and recently it has been shown that sulindac and its metabolites inhibited multiple transport proteins in primary rat and human hepatocytes as tested with model substrates, e.g., taurocholate and estradiol-17β-glucuronide (Lee et al., 2010). To date, the molecular mechanisms of NSAID uptake into hepatocytes are largely unknown. Furthermore, no in vitro studies investigating hepatic uptake transporter-mediated drug-drug interactions between NSAIDs and other drugs are available.
Therefore, we investigated the influence of the NSAIDs acetylsalicylic acid, diclofenac, ibuprofen, lumiracoxib, naproxen, and salicylic acid and of paracetamol on OATP1B1- and OATP1B3-mediated uptake of the prototypic substrate bromosulfophthalein (BSP) and of the widely prescribed HMG-CoA reductase inhibitor pravastatin and tested whether NSAIDs are transport substrates for these uptake transporters. Furthermore, the effect of ibuprofen, diclofenac, and lumiracoxib on OATP1B1- and OATP1B3-mediated pravastatin transport kinetics was investigated in detail, demonstrating that these NSAIDs influenced OATP1B1- and OATP1B3-mediated transport kinetics of pravastatin.
Materials and Methods
Chemicals.
Ibuprofen was purchased from Biomol GmbH (Hamburg, Germany). Pravastatin sodium was obtained from BioTrend GmbH (Wangen/Zurich, Switzerland). Acetylsalicylic acid, salicylic acid, paracetamol, diclofenac sodium, and naproxen were purchased from Sigma-Aldrich (Taufkirchen, Germany). Lumiracoxib was obtained from Chemos GmbH (Regenstauf, Germany). Acetylsalicylic acid, salicylic acid, paracetamol, diclofenac sodium, ibuprofen, lumiracoxib, naproxen, and pravastatin sodium were dissolved in dimethyl sulfoxide. MTT was obtained from Invitrogen GmbH (Karlsruhe, Germany). Acetonitrile hypergrade for LC-mass spectrometry was purchased from Merck (Darmstadt, Germany) and ammonium acetate was obtained from Sigma-Aldrich. [14C]Diclofenac (55 mCi/mmol), [14C]ibuprofen (55 mCi/mmol), [14C]naproxen (50 mCi/mmol), and [14C]salicylic acid (55 mCi/mmol) were obtained from American Radiolabeled Chemicals (St. Louis, MO). [3H]Sulfobromophthalein was obtained from Hartmann Analytic (Braunschweig, Germany). All other chemicals and reagents, unless stated otherwise, were obtained from Carl Roth GmbH + Co. KG (Karlsruhe, Germany).
Cell Culture.
Human embryonic kidney (HEK293) cells and Madin-Darby canine kidney (MDCKII) cells were cultured in minimal essential medium containing 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. The cells were routinely subcultured by trypsinization using a trypsin (0.05%)-EDTA (0.02%) solution. All cell culture media supplements were obtained from Invitrogen GmbH (Karlsruhe, Germany).
Uptake Studies.
HEK-OATP1B1, HEK-OATP1B3, MDCK-OATP1B1, and MDCK-OATP1B3 cells and the respective vector control cells HEK293-VC G418, HEK293-VC Hygro, and MDCK-VC G418 were used as described previously (König et al., 2000a,b; Seithel et al., 2007). In brief, 12-well plates were coated with poly-d-lysine (Greiner Bio-One, Frickenhausen, Germany). Cells were seeded in these plates at a density of 7 × 105 cells/well. After 24 h, cells were incubated for 24 h with 10 mM sodium butyrate medium to increase protein expression (Cui et al., 1999). Before the uptake experiment, cells were washed with prewarmed (37°C) uptake buffer (142 mM NaCl, 5 mM KCl, 1 mM K2PO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM glucose, and 12.5 mM HEPES, pH 7.3). Subsequently, cells were incubated with the test solution, containing the substrate pravastatin or BSP or/and the NSAID/paracetamol, at 37°C for 10 min. Over this time period, linearity of uptake has been demonstrated (Hsiang et al., 1999; König et al., 2000b). Then cells were washed three times with ice-cold uptake buffer and lysed with 0.2% SDS. Intracellular accumulation of pravastatin was determined using a LC-MS/MS method (Seithel et al., 2007) as described in detail later. Intracellular accumulation of radioactivity was measured by liquid scintillation counting (TriCarb 2800; Perkin Elmer Life and Analytical Sciences, Bonn, Germany). The protein concentration of each sample was determined using a bicinchoninic acid assay (BCA Protein Assay Kit; Thermo Fisher Scientific, Waltham, MA). All experiments were performed with HEK cell lines described above. To exclude cell line-specific effects, the impact of ibuprofen on pravastatin uptake was also investigated in the MDCK cell lines described above (data not shown).
Cytotoxicity Assay.
To assess cell viability, a MTT assay was used. Experiments were performed as described for the uptake experiments. After incubation with the test solution, cells were washed three times with uptake buffer at 37°C. Subsequently, cells were incubated with 50 μl of MTT solution (5 mg/ml) in 500 μl of medium for 3 h. After that, the medium was removed, and cells were lysed with 800 μl of a mixture of 0.6% HCl and 10% SDS in dimethyl sulfoxide. The absorption was read at 570 nm, and 70% ethanol-H2O (v/v) was used as a positive control.
LC-MS/MS Assay for Pravastatin.
Pravastatin measurements were performed by LC-MS/MS as described previously (Seithel et al., 2007), with minor modifications. In brief, the samples were diluted with mobile phase or internal standard (ibuprofen) and injected directly. The first and the last minute of each run were discarded through a valve (VICI Valco Instrument Co. Inc., Houston, TX) to minimize matrix effects. Because of an increase in pressure over time with use of a column with a 3-μm particle size, a Nucleosil 100-5 C18 AB column (4 mm × 125 mm, particle size 5 μm; Machery-Nagel, Düren, Germany) with a corresponding guard column (3 mm × 8 mm, particle size 5 μm; Machery-Nagel) was applied instead. The mobile phase was changed to a mixture of 12 mM ammonium acetate and acetonitrile (50:50; v/v) to sharpen the peak form. The LC-MS/MS system consisted of an API 4000 mass spectrometer (Applied Biosystems, Darmstadt, Germany) equipped with an Agilent 1100 HPLC System (Agilent Technologies, Waldbronn, Germany). Standards for the calibration curve were freshly prepared before each run. Samples containing ibuprofen as inhibitor were quantified over an external calibration curve.
Data Analysis.
The OATP1B1- and OATP1B3-mediated net uptake was obtained by subtracting the uptake in vector-transfected cells from that in OATP1B1- and OATP1B3-expressing cells. The corresponding EC50 values for stimulation of OATP1B1- and OATP1B3-mediated pravastatin uptake were calculated by fitting the data to a one-site binding (hyperbola) nonlinear regression curve (Prism 4.01 2004; GraphPad Software Inc., San Diego, CA). The EC50 value is the concentration at which 50% stimulation of substrate uptake is obtained. Transport kinetics were calculated by Prism 4.01 2004. The calculated parameters were the Michaelis-Menten constant (Km) and maximum transport rate (Vmax).
Statistical Analysis.
Uptake, inhibition, and stimulation were determined in two independent experiments on separate days with three wells per concentration and day (unless stated otherwise); i.e., for any concentration six separate wells were investigated. All data are presented as means ± S.E.M. Multiple comparisons were analyzed by analysis of variance with a subsequent Dunnett multiple comparison test or Bonferroni post-test. P < 0.05 was required for statistical significance.
Results
Influence of NSAIDs and Paracetamol on OATP1B1- and OATP1B3-Mediated BSP Uptake.
First we investigated whether NSAIDs and paracetamol (for pharmacokinetic data, see Table 1) interact with OATP1B1- and OATP1B3-mediated uptake. For this we used BSP as the prototypical substrate under standardized uptake conditions (Seithel et al., 2007). Selected NSAIDs or paracetamol were added in two concentrations (10 and 50 μM). As shown in Fig. 1, diclofenac at a concentration of 50 μM, ibuprofen at a concentration of 10 μM, and lumiracoxib at both tested concentrations showed significant inhibition of OATP1B1-mediated BSP uptake. Significant inhibition of OATP1B3-mediated BSP uptake was detected for diclofenac at a concentration of 50 μM, for ibuprofen at a concentration of 10 μM, and for lumiracoxib at a concentration of 50 μM.
Pharmacokinetic data for NSAIDs and paracetamol

Influence of acetylsalicylic acid (ASA), diclofenac (Diclo), ibuprofen (Ibu), lumiracoxib (Lumira), naproxen (Napro), paracetamol (PCM), and salicylic acid (SA) on OATP1B1-mediated (top) and OATP1B3-mediated (bottom) BSP (0.05 μM for OATP1B1; 1 μM for OATP1B3) uptake. Uptake was measured for 10 min. Data are from two independent experiments with a total of six wells per concentration and are shown as a percentage of BSP uptake in the absence of the respective drug. Each value is the mean value ± S.E.M. *, P < 0.05 versus control; **, P < 0.01 versus control.
Influence of NSAIDs and Paracetamol on OATP1B1- and OATP1B3-Mediated Pravastatin Uptake.
Because BSP is not a drug substrate we next investigated the influence of the same NSAIDs and of paracetamol on OATP1B1- and OATP1B3-mediated pravastatin uptake (Fig. 2). BSP and pravastatin are both substrates for OATP1B1 and OATP1B3 and BSP inhibited OATP1B1- and OATP1B3-mediated pravastatin uptake with Ki values of 85 and 900 nM, respectively (data not shown). OATP1B1-mediated pravastatin uptake was significantly stimulated by both concentrations of diclofenac, by ibuprofen at a concentration of 50 μM, by lumiracoxib at a concentration of 10 μM, and by naproxen at a concentration of 50 μM. Significant stimulation of OATP1B3-mediated pravastatin uptake was detected for diclofenac at both concentrations (10 and 50 μM) and for ibuprofen at a concentration of 50 μM. In contrast to OATP1B1- and OATP1B3-mediated BSP uptake, no significant inhibition of pravastatin uptake could be detected.

Influence of acetylsalicylic acid (ASA), diclofenac (Diclo), ibuprofen (Ibu), lumiracoxib (Lumira), naproxen (Napro), paracetamol (PCM), and salicylic acid (SA) on OATP1B1-mediated (top) and OATP1B3-mediated (bottom) pravastatin (50 μM) uptake. Uptake was performed for 10 min. Data are from two independent experiments with a total of six wells per concentration and are shown as a percentage of pravastatin uptake in the absence of the respective drug. Each value is the mean value ± S.E.M. *, P < 0.05 versus control; **, P < 0.01 versus control.
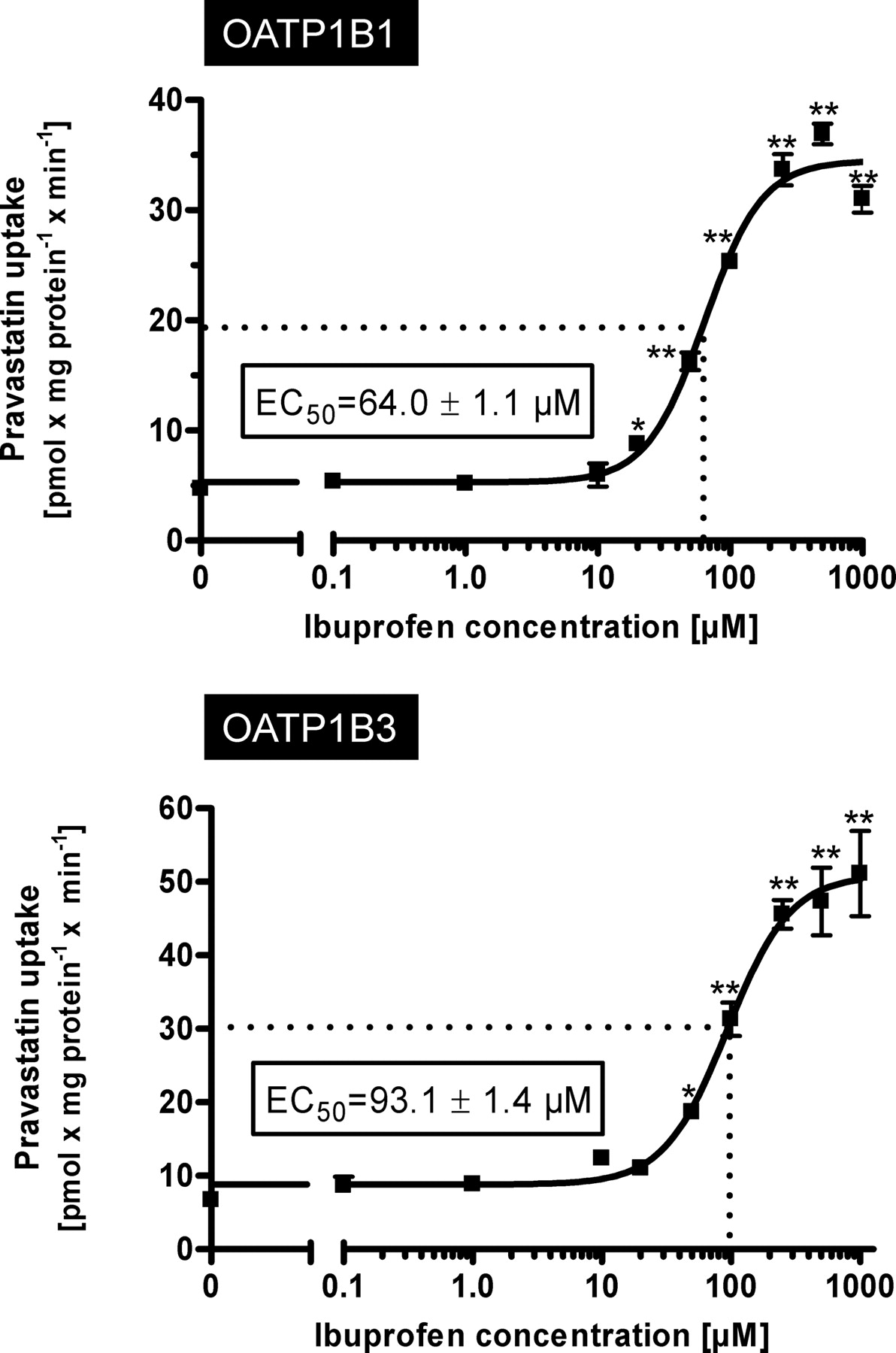
Stimulation of OATP1B1- and OATP1B3-Mediated Pravastatin Uptake by Ibuprofen.
To further investigate this stimulatory effect on pravastatin uptake, we performed concentration dependence experiments using pravastatin at a concentration of 50 μM as the substrate and increasing concentrations of ibuprofen as the interacting drug (Fig. 3). These experiments revealed EC50 values of 64.0 μM for OATP1B1-mediated pravastatin uptake and 93.1 μM for OATP1B3-mediated pravastatin uptake.

Kinetics of OATP1B1-mediated (top) and OATP1B3-mediated (bottom) uptake of pravastatin (50 μM) dependent on ibuprofen concentration. Net uptake was calculated as the difference between pravastatin uptake into OATP1B1- and OATP1B3-expressing cells and uptake into the respective vector control cells. Uptake was measured for 10 min. Each value is the mean value from one experiment with a total of three wells per concentration ± S.E.M. *, P < 0.05 versus control; **, P < 0.01 versus control.
To exclude cell-specific effects based on stably transfected HEK293 cells, these stimulation experiments were repeated using stably transfected MDCKII cells recombinantly overexpressing OAP1B1 and OATP1B3 and the respective vector control cells (König et al., 2000a,b). These experiments revealed comparable results, also demonstrating a significant increase in OATP1B1- and OATP1B3-mediated pravastatin uptake by addition of increased concentrations of ibuprofen (data not shown).
Furthermore, to exclude cytotoxic effects, cell viability was tested by an MTT assay. No cytotoxic effect of ibuprofen added in very high concentrations (up to 1000 μM) was demonstrated.
Influence of Ibuprofen on OATP1B1- and OATP1B3-Mediated Pravastatin Uptake Kinetics.
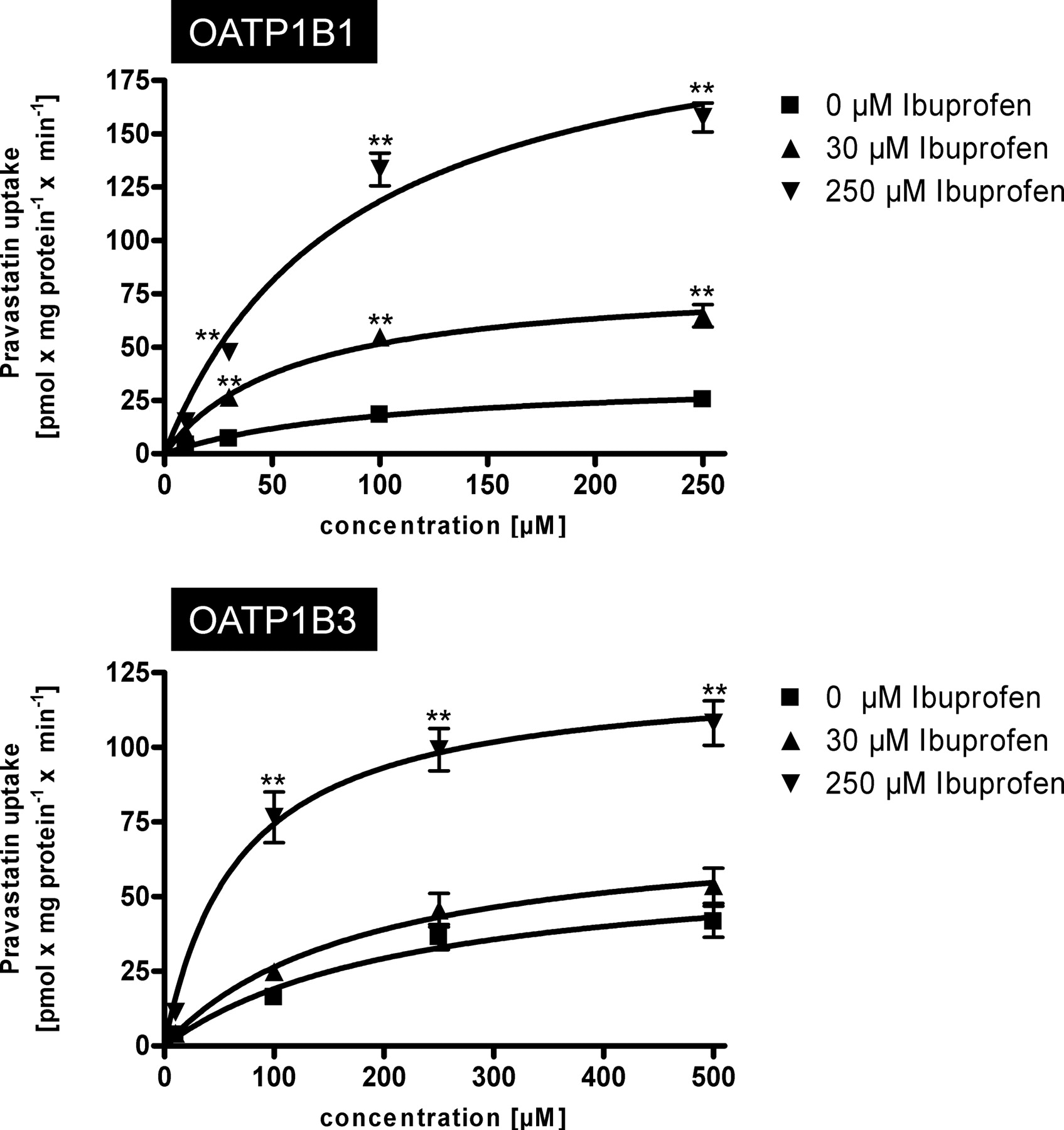
A detailed kinetic analysis of the effect of ibuprofen on OATP1B1- and OATP1B3-mediated pravastatin uptake kinetics is shown in Fig. 4. For OATP1B1, 30 and 250 μM ibuprofen increased the Vmax value from 36.3 to 82.3 and 221.2 pmol × mg protein−1 × min−1, respectively whereas the Km value decreased from 104.0 to 59.7 and 86.8 μM, respectively (Table 2). The same effect could be detected for OATP1B3-mediated pravastatin uptake (Table 2). The Vmax value increased from 62.8 to 74.7 and 124.8 pmol × mg protein−1 × min−1, respectively, whereas the Km value decreased from 228.0 to 182.7 and 67.8 μM, respectively.
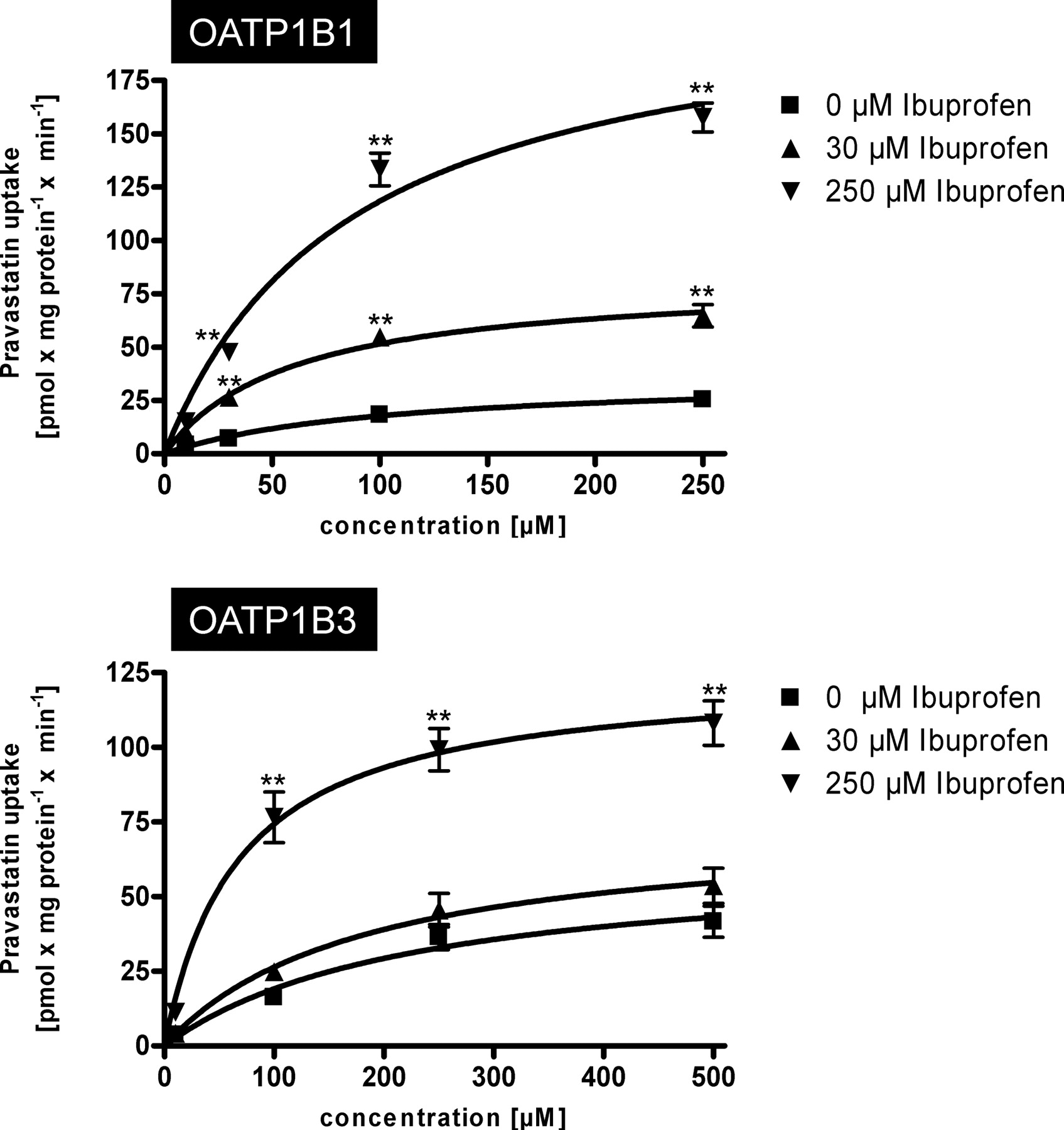
Kinetics of OATP1B1-mediated (top) and OATP1B3-mediated (bottom) uptake of pravastatin without and in presence of 30 and 250 μM ibuprofen. Net uptake was calculated as the difference between pravastatin uptake into OATP1B1- and OATP1B3-expressing cells and uptake into the respective vector control cells. Uptake was measured for 10 min. Each value is the mean value from two independent experiments with a total of six wells per concentration ± S.E.M. **, P < 0.01 versus control.
Comparison of OATP1B1- and OATP1B3-mediated pravastatin uptake kinetics in the absence or presence of ibuprofen
Km and Vmax data are derived from data shown in Fig. 4.
Influence of Diclofenac and Lumiracoxib on OATP1B1- and OATP1B3-Mediated Pravastatin Uptake.
Whereas ibuprofen stimulated OATP1B1- and OATP1B3-mediated pravastatin uptake at high concentrations also, diclofenac and lumiracoxib showed the highest stimulation at a low NSAID concentration (10 μM) and at the higher concentration (50 μM) stimulation was reduced (Fig. 2). Therefore, we performed concentration dependence experiments with added diclofenac and lumiracoxib concentrations between 0.1 and 250 μM. These experiments demonstrated that diclofenac increased OATP1B1- and OATP1B3-mediated pravastatin uptake up to 4.2- and 2.5-fold at a concentration of 10 μM, respectively, whereas higher diclofenac concentrations showed reduced stimulation. At the highest tested diclofenac concentration (250 μM), OATP1B3-mediated uptake was reduced to 19%, whereas no uptake inhibition could be observed for OATP1B1-mediated pravastatin transport. Experiments with lumiracoxib revealed similar results. At a concentration of 10 μM, lumiracoxib OATP1B1- and OATP1B3-mediated pravastatin transport was significantly increased 2.6- and 1.2-fold, respectively. Higher concentrations of lumiracoxib led to lower intracellular accumulation of pravastatin compared with that of 10 μM lumiracoxib (Fig. 5). For lumiracoxib, no pravastatin uptake inhibition was observed. Furthermore, the detailed kinetic analysis of the effects of diclofenac and lumiracoxib using the same pravastatin concentrations as presented in Fig. 4 demonstrated that for OATP1B1 10 μM diclofenac or lumiracoxib increased the Km value from 104.0 to 135.1 and 317.7 μM, respectively. Under the same conditions, the Vmax value increased from 36.3 to 85.8 and 147.5 pmol × mg protein−1 × min−1, respectively (Table 3). Different effects on OATP1B3-mediated pravastatin uptake could be observed for these two NSAIDs used in the same concentration of 10 μM. The Km value decreased from 228.0 to 24.3 μM in the presence of diclofenac and to 52.4 μM in the presence of lumiracoxib, and the Vmax value decreased from 62.8 to 15.9 and 29.0 pmol × mg protein−1 × min−1, respectively.
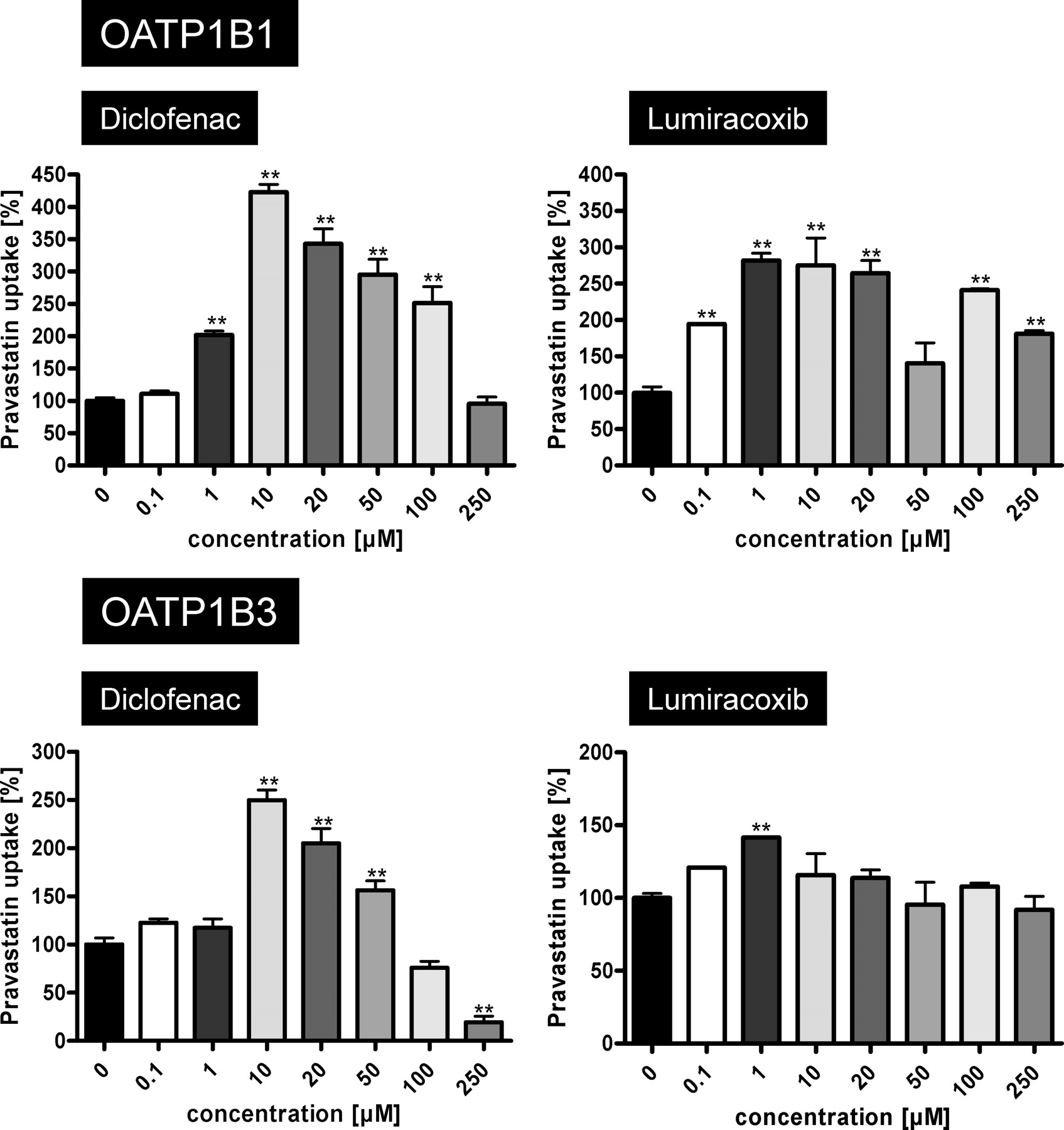
OATP1B1-mediated (top) and OATP1B3-mediated (bottom) uptake of pravastatin (50 μM) in dependence on diclofenac and lumiracoxib concentrations. Net uptake was calculated as the difference between pravastatin uptake into OATP1B1- and OATP1B3-expressing cells and uptake into the respective vector control cells. Uptake was measured for 10 min. Each value is the mean value of one experiment with a total of three wells per concentration ± S.E.M. **, P < 0.01 versus control.
Comparison of OATP1B1- and OATP1B3-mediated pravastatin uptake kinetics in the absence and presence of the NSAIDs diclofenac and lumiracoxib
OATP1B1- and OATP1B3-Mediated Uptake of Diclofenac, Ibuprofen, Naproxen, and Salicylic Acid.
Finally, labeled diclofenac, ibuprofen, naproxen, and salicylic acid were tested as substrates for OATP1B1 and OATP1B3. A modest, but significant, uptake could be detected only for OATP1B3-mediated diclofenac uptake at concentrations of 1, 10, and 100 μM (Fig. 6). No significantly increased uptake of the other NSAIDs could be detected in OATP1B1- or OATP1B3-expressing cells compared with the uptake into vector control cells (data not shown).
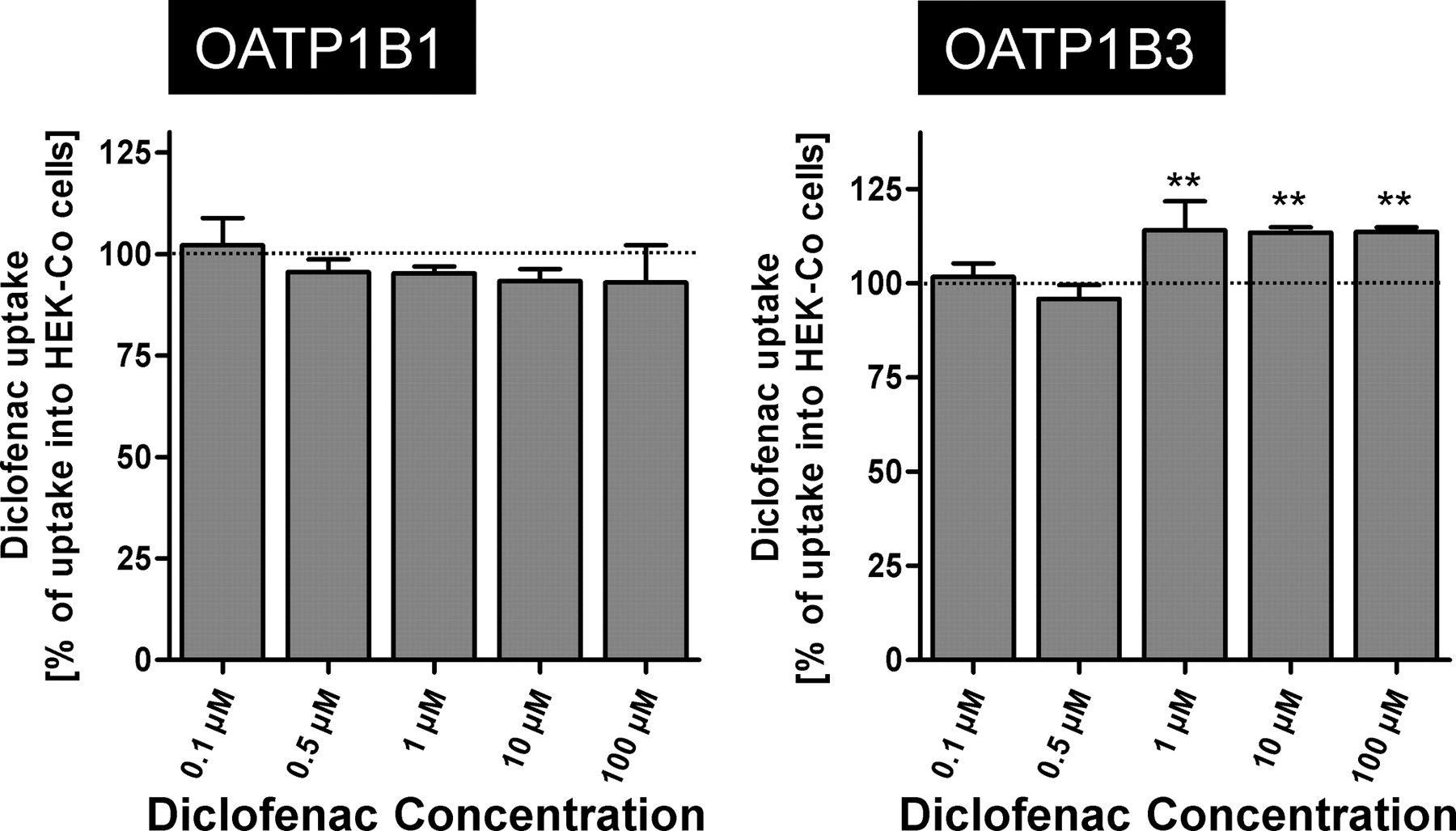
OATP1B1-mediated (left) and OATP1B3-mediated (right) uptake of diclofenac. Data are shown as a percentage of NSAID uptake into the vector control cells. Uptake was measured for 10 min. Each value is the mean value from one experiment with a total of three wells per concentration ± S.E.M. **, P < 0.01 versus control.
Discussion
In this study, we investigated the influence of several widely prescribed NSAIDs and of paracetamol on the hepatic drug uptake mediated by the OATP family members OATP1B1 and OATP1B3. Drug uptake transporters are important determinants of drug pharmacokinetics and alterations in drug uptake rates, e.g., by transporter-mediated drug-drug interactions, may result in a change in drug pharmacokinetics with the risk of adverse drug reactions. In the case of statins as drug transporter substrates, several studies have analyzed in vitro and in vivo the effect of coadministered drugs influencing OATP-mediated statin uptake into hepatocytes. Cyclosporine, for instance, inhibited OATP1B1- and OATP1B3-mediated drug uptake in vitro (Treiber et al., 2007). Thus, it has been observed in humans that coadministration of the OATP1B1 and OATP1B3 substrate pravastatin with cyclosporine increased the plasma concentrations of pravastatin (Regazzi et al., 1993; Olbricht et al., 1997; Park et al., 2002; Hedman et al., 2004). Because pravastatin is not metabolized to a significant extent (Jacobsen et al., 1999), inhibition of OATP1B1- and OATP1B3-mediated pravastatin uptake may be one relevant molecular mechanism behind this observed drug-drug interaction. So far, several NSAIDs including indomethacin, ibuprofen, ketoprofen, and naproxen have been identified as inhibitors of rat Oatp1a1 and rat Oatp1a3 (Shitara et al., 2002), but the influence of NSAIDs and of paracetamol on human OATP-mediated drug uptake has not been investigated so far. In addition, because most of the widely used NSAIDs and paracetamol are metabolized in liver before renal or biliary excretion (Brater, 1988), transporter-mediated uptake of these drugs may be important for disposition of NSAIDs and paracetamol.
For these reasons, we investigated whether paracetamol and the NSAIDs acetylsalicylic acid, diclofenac, ibuprofen, lumiracoxib, naproxen, and salicylic acid interact with OATP1B1- and OATP1B3-mediated transport. Expected maximal plasma concentrations and predicted total concentrations in the portal venous blood of these NSAIDs are summarized in Table 1. In a first approach we used BSP as a standard substrate, demonstrating that diclofenac, ibuprofen, and lumiracoxib inhibited BSP uptake, which suggests that some but not all NSAIDs we investigated may interact with these uptake transporters (Fig. 1). This interaction was subsequently investigated using pravastatin as drug substrate for both hepatic OATPs, but interestingly, even when diclofenac, ibuprofen, and lumiracoxib were used in the same concentrations, no inhibition but stimulation of pravastatin uptake was observed (Fig. 2). For BSP and pravastatin, acetylsalicylic acid, paracetamol, and salicylic acid showed neither uptake inhibition or stimulation. For acetylsalicylic acid, this finding is in line with a single-dose crossover study in healthy volunteers in which no pharmacokinetic interaction of orally administered acetylsalicylic acid with pravastatin was observed (PRAVIGARD PAC product information, 2006; Bristol-Myers Squibb, Princeton, NJ).
In most cases of transporter-mediated drug interactions, uptake of one drug is dose-dependently inhibited by the added interacting second drug. This observation suggests that the second interacting drug is also a substrate for the uptake transporter and that reduced uptake of the first drug is due to the competitive inhibition. In our study, uptake inhibition could be observed for BSP as substrate and for high concentrations of diclofenac (Fig. 1), whereas for pravastatin as substrate diclofenac at both concentrations stimulated pravastatin uptake. Strong pravastatin uptake stimulation was also observed for ibuprofen, whereas only a slight BSP uptake inhibition was detected. This result suggests an allosteric mechanism, in which the NSAID binds to the transporter, altering the affinity and transport kinetics for BSP and for pravastatin without competing for drug transport. This hypothesis was supported by the kinetic analysis (Fig. 4; Tables 2 and 3), for which we have found that both kinetic values, the kinetic constant Km as well as the maximal transport velocity Vmax, were altered by addition of ibuprofen, diclofenac, or lumiracoxib to the pravastatin uptake assay. In the case of competitive inhibition, only the Km value should be altered. This observation suggests that these NSAIDs bind to an allosteric interaction site modulating the transport kinetics for OATP1B1- and OATP1B3-mediated pravastatin uptake by an allosteric mechanism without being transported themselves. Furthermore, this allosteric mechanism could explain why ibuprofen and the other NSAIDs were not transported by OATP1B1 and OATP1B3 (Fig. 6), whereas diclofenac shows slight but significant transport by OATP1B3 and inhibits OATP1B3-mediated pravastatin uptake at high concentrations. OATP1B3-mediated diclofenac uptake was observed only at higher diclofenac concentrations (>1 μM), suggesting that the affinity of OATP1B3 for this NSAID is low and, therefore, higher diclofenac concentrations are necessary for significant uptake into HEK-OATP1B3 cells compared with the uptake into HEK control cells, mediated by endogenous uptake transporters. This allosteric transport modulation could also explain why diclofenac, ibuprofen, and lumiracoxib inhibited BSP uptake without activating it at low NSAID concentrations. Of interest, these different effects of substrates or drugs on OATP-mediated transport have been published before. Gui et al. (2008) found that clotrimazole stimulated OATP1B3-mediated estradiol-17β-glucuronide uptake but inhibited the transport of Fluo-3. OATP1B1- and OATP1B3-mediated pravastatin uptake stimulation at low drug concentrations has also been demonstrated for the oral antidiabetes drug rosiglitazone, whereas at high rosiglitazone concentrations pravastatin uptake was inhibited (Bachmakov et al., 2008).
Different binding or interacting sites on OATPs have been described also for the third hepatic OATP family member OATP2B1. In a study using estrone-3-sulfate as substrate, it was demonstrated that the transport follows biphasic uptake kinetics with a high-affinity component (Km value of 1.8 μM) and a low-affinity component (Km value of 1.4 mM) (Sai et al., 2006). This biphasic uptake kinetics was also shown for OATP1B1-mediated estrogen 3-sulfate uptake (Noé et al., 2007), and it was demonstrated that only the high-affinity component was inhibited by gemfibrozil. These findings together with our experiments presented here demonstrate that hepatic OATP family members have different binding sites for (drug) substrates and that binding of a (drug) substrate to one site might allosterically alter the uptake kinetics for a second drug (substrate).
The clinical consequences of such stimulatory effects are difficult to predict. Clinical trials investigating potential drug-drug interactions between NSAIDs and pravastatin are largely lacking. Stimulation of hepatocellular pravastatin uptake could have a benefit in leading to higher intracellular pravastatin concentrations and therefore to a more pronounced inhibition of the target enzyme HMG-CoA reductase.
In summary, we have demonstrated that NSAIDs interact with the hepatic uptake transporters OATP1B1 and OATP1B3 by altering the uptake kinetics for the standard substrate BSP and the HMG-CoA reductase inhibitor pravastatin. Whereas BSP uptake was inhibited by some NSAIDs, pravastatin uptake was stimulated by the same NSAIDs. Only diclofenac was significantly transported by OATP1B3, whereas the other NSAIDs were not substrates for OATP1B1 and OATP1B3. These results demonstrated that, in addition to competitive uptake inhibition, other kinetic mechanisms such as allosteric activation may play a role in transporter-mediated drug-drug interactions.
Authorship Contributions
Participated in research design: Kindla, Müller, Fromm, and König.
Conducted experiments: Kindla, Müller, and Mieth.
Contributed new reagents or analytic tools: Mieth.
Performed data analysis: Kindla, Müller, and Mieth.
Wrote or contributed to the writing of the manuscript: Kindla, Müller, Mieth, Fromm, and König.
Footnotes
This work was supported by the Deutsche Krebshilfe [Grant 107854]; and the Deutsche Forschungsgemeinschaft [Grant DFG Ko 2120/1-3].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.037622.
-
ABBREVIATIONS:
- SLC
- solute carrier
- OATP/Oatp
- organic anion transporting polypeptide
- NSAID
- non-steroidal anti-inflammatory drug
- BSP
- sulfobromophthalein
- MTT
- 3(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LC
- liquid chromatography
- HEK
- human embryonic kidney
- MDCK
- Madin-Darby canine kidney
- MS/MS
- tandem mass spectrometry.
- Received December 7, 2010.
- Accepted March 9, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}