


Abstract
For dual transporter-enzyme substrate drugs, the extended clearance model can be used to predict the rate-determining step(s) (RDS) of a drug and hence predict its drug-drug interaction (DDI) liabilities (i.e., transport, metabolism, or both). If the RDS of the hepatic clearance of the drug is sinusoidal uptake clearance (CLsin), even if the drug is eliminated mainly by hepatic metabolism, its DDI liability (as viewed from changes to systemic drug concentrations) is expected to be inhibition or induction of uptake transporters but not hepatic enzymes; however, this is true only if the condition required to maintain CLsin as the RDS is maintained. Here, we illustrate through theoretical simulations that the RDS condition may be violated in the presence of a DDI. That is, the RDS of a drug can switch from CLsin to all hepatobiliary clearances [i.e., metabolic/biliary clearance (CLmet + bile) and CLsin], leading to unexpected systemic DDIs, such as metabolic DDIs, when only transporter DDIs were anticipated. As expected, these analyses revealed that the RDS switch depends on the ratio of CLmet + bile to sinusoidal efflux clearance (CLsef). Additional analyses revealed that for intravenously administered drugs, the RDS switch also depends on the magnitude of CLsin. We analyzed published in vitro quantified hepatobiliary clearances and observed that most drugs have a CLmet + bile/CLsef ratio < 4; hence, in practice, the magnitude of CLsin must be considered when establishing the RDS. These analyses provide insights previously not appreciated and a theoretical framework to predict DDI liabilities for drugs that are dual transporter-enzyme substrates.
Introduction
Identifying liabilities with respect to drug-drug interactions (DDI) is important in drug development. In 2015, 25 of the 33 new drug applications contained in vitro transporter data, and of 20 clinical trials using the new molecular entities (NMEs) as victim drugs, only nine resulted in a significant area under the curve (AUC) change (Yu et al., 2017). These data acknowledge that drug transporters are important in determining drug disposition (Giacomini et al., 2010; Hillgren et al., 2013; Patel et al., 2016).
As shown by the hepatic extended clearance model (ECM), when a drug is both transported into and metabolized or biliary-excreted by the liver, the rate-determining step (RDS) in the systemic clearance of the drug can be its hepatic uptake clearance, metabolic clearance, biliary (canalicular efflux) clearance, or all hepatobiliary clearances (Miyauchi et al., 1987; Sirianni and Pang, 1997; Shitara et al., 2006; Kusuhara and Sugiyama, 2009; Li et al., 2014; Patilea-Vrana and Unadkat, 2016). The RDS of a drug can be identified using models such as the Extended Clearance Concept Classification System and the Extended Clearance Classification System, which use the drug’s in vitro quantified hepatobiliary clearance values or the drug’s physicochemical properties, respectively (Camenisch and Umehara, 2012; Varma et al., 2015). Using such models is advantageous since the RDS of a drug helps identify where the DDI liabilities lie. Of note, unless indicated otherwise, all subsequent references to DDI should be interpreted as those DDIs that can be observed from measurement of the systemic concentrations of the victim drug. For example, if the RDS of a drug is its hepatic uptake clearance (RDSuptake), then the focus of the DDI studies should be transporter-based [e.g., hepatic organic anion-transporting polypeptide (OATP)–mediated uptake of atorvastatin] (Maeda et al., 2011) or if the RDS is both hepatic uptake and metabolic or biliary clearance (RDSall), the focus of DDI studies should be all hepatobiliary pathways (e.g., OATP and cytochrome P450-mediated clearance of cerivastatin) (Mück et al., 1999; Backman et al., 2002).
Here, we asked whether knowledge of the RDS of a drug is enough to predict DDI liabilities for drugs that are dual transporter-enzyme substrates. If not, the focus of DDI studies will be misdirected and will result in either a negative or unexpected DDI and therefore toxicity. Under the worst-case scenario, the latter will lead to discontinuation of drug development and the end result is that both outcomes will increase drug development cost (Paul et al., 2010). For these reasons, it is important to ask whether the RDS can switch from hepatic uptake clearance to all hepatobiliary clearance pathways thus resulting in unexpected systemic DDIs. Using the ECM theory and simulations, we aimed to: 1) provide a theoretical framework of when the RDSuptake switches to RDSall in the presence of a DDI and 2) apply the RDS framework to predict DDI liabilities through theoretical and practical examples. The resulting analyses and simulations provide novel insights, hitherto not appreciated, into factors that determine when a victim drug experiences the RDSuptake switch to RDSall and elucidate important considerations for predicting DDI liabilities for drugs that are substrates of both hepatic transporters and enzymes.
Materials and Methods
Theoretical Background.
The ECM describes complex hepatobiliary clearance in terms of transport at the sinusoidal membrane via sinusoidal influx (CLsin) and efflux (CLsef), transport at the canalicular membrane via biliary efflux (CLbile), metabolism (CLmet), hepatic blood flow (Qh), and fraction unbound in blood (fub) (eq. 1). CLsin and CLsef terms incorporate both transport-mediated plus passive diffusion clearance, whereas CLbile describes active transport only. The interrelationships between the hepatobiliary clearances defined by the ECM create the RDS in the hepatic clearance of a drug. As described by us and others (Miyauchi et al., 1987; Sirianni and Pang, 1997; Shitara et al., 2006; Patilea-Vrana and Unadkat, 2016), these can be 1) RDSmet + bile when the metabolic and biliary efflux clearances of the drug are much less than sinusoidal efflux clearance (CLmet + bile < < CLsef) and the drug is highly permeable (passive diffusion > > active transport, CLsin ≈ CLsef) and thus can rapidly distribute across the sinusoidal membrane; 2) RDSuptake when the metabolic plus biliary efflux clearances are much greater than the sinusoidal efflux clearance (CLmet + bile > > CLsef), or 3) RDSall when a drug has both active transport and metabolism, but the preceding two extreme scenarios do not apply (CLsin ≠ CLsef):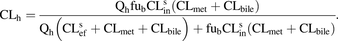 (1)Identifying the RDS of a drug can be used to predict the liability of transporter versus metabolic DDIs (see Patilea-Vrana and Unadkat, 2016, for simulations of systemic and hepatic AUC when hepatobiliary clearances are inhibited). For example, although a victim drug has RDSuptake, inhibition of CLmet + bile will not result in a significant increase in the systemic AUC, even though such DDI could result in significant drug accumulation in the liver and hence potentially enhance the hepatic efficacy or toxicity of the drug. That is, from the point of view of systemic (e.g., victim plasma concentrations) measurements, inhibition of CLmet + bile will be incorrectly interpreted as negative because there will be no change in systemic concentrations of the drug. On the other hand, inhibition of CLsin will result in an increase in the drug’s systemic AUC (and therefore potentially nonhepatic efficacy and toxicity of the drug) but will result in no changes in the hepatic AUC, provided the liver is the primary eliminating organ (for examples, see Patilea-Vrana and Unadkat, 2016). Less appreciated, however, is the fact that in the presence of metabolic or biliary efflux DDI, the RDS of a drug can switch from RDSuptake to RDSall and hence switch the DDI liability from uptake transporters to both metabolic or biliary and uptake pathways. Consequently, the drug’s systemic AUC will significantly change owing to metabolic and biliary efflux DDIs, even though uptake was the RDS of the drug in the absence of a DDI, which would lead to unexpected DDIs as viewed from the systemic concentrations of the victim drug. Therefore, through MATLAB simulations (R2016a; MathWorks, Natick, MA), we illustrated when the RDSuptake-to-RDSall switch occurs for a victim drug in the presence of a DDI. We then applied our proposed RDS framework to published in vitro hepatobiliary clearances to determine whether in vivo observed DDI liabilities can be correctly predicted. Although the insights illustrated can be derived from analytical solutions of the ECM equation (eq. 1), for clarity, we chose to use simulations to illustrate the principles of these DDI liabilities within the RDS framework.
(1)Identifying the RDS of a drug can be used to predict the liability of transporter versus metabolic DDIs (see Patilea-Vrana and Unadkat, 2016, for simulations of systemic and hepatic AUC when hepatobiliary clearances are inhibited). For example, although a victim drug has RDSuptake, inhibition of CLmet + bile will not result in a significant increase in the systemic AUC, even though such DDI could result in significant drug accumulation in the liver and hence potentially enhance the hepatic efficacy or toxicity of the drug. That is, from the point of view of systemic (e.g., victim plasma concentrations) measurements, inhibition of CLmet + bile will be incorrectly interpreted as negative because there will be no change in systemic concentrations of the drug. On the other hand, inhibition of CLsin will result in an increase in the drug’s systemic AUC (and therefore potentially nonhepatic efficacy and toxicity of the drug) but will result in no changes in the hepatic AUC, provided the liver is the primary eliminating organ (for examples, see Patilea-Vrana and Unadkat, 2016). Less appreciated, however, is the fact that in the presence of metabolic or biliary efflux DDI, the RDS of a drug can switch from RDSuptake to RDSall and hence switch the DDI liability from uptake transporters to both metabolic or biliary and uptake pathways. Consequently, the drug’s systemic AUC will significantly change owing to metabolic and biliary efflux DDIs, even though uptake was the RDS of the drug in the absence of a DDI, which would lead to unexpected DDIs as viewed from the systemic concentrations of the victim drug. Therefore, through MATLAB simulations (R2016a; MathWorks, Natick, MA), we illustrated when the RDSuptake-to-RDSall switch occurs for a victim drug in the presence of a DDI. We then applied our proposed RDS framework to published in vitro hepatobiliary clearances to determine whether in vivo observed DDI liabilities can be correctly predicted. Although the insights illustrated can be derived from analytical solutions of the ECM equation (eq. 1), for clarity, we chose to use simulations to illustrate the principles of these DDI liabilities within the RDS framework.
Simulation Assumptions.
The hepatic ECM was simulated using the governing differential equations as previously described (Endres et al., 2009; Patilea-Vrana and Unadkat, 2016), and for simplicity, the following assumptions about the victim drug were made: 1) it was administered intravenously; 2) the fraction unbound (fu) in blood and tissue (liver) was set to 1; 3) liver was the only eliminating organ; 4) Qh was set to 1 liter/min. All references to systemic AUC are derived from drug concentrations in blood. Our conclusions regarding the RDS switch are generalizable to when victim drugs are administered orally, but our conclusions of the RDS dependence on CLsin apply only to intravenously administered drugs (see text to follow). Furthermore, for oral drug administration, our findings apply only to changes to the hepatic clearance and bioavailability of the victim drug and do not address the intestinal availability of the victim drug. If there is significant nonhepatic clearance, our conclusions will stand except that the magnitude of the change observed in the systemic and/or hepatic AUC of the drug will differ (Patilea-Vrana and Unadkat, 2016).
Identifying When the RDSuptake Switches to RDSall and Factors that Influence this Switch.
First, we determined when the RDS of a drug switches from uptake clearance to all hepatobiliary clearance pathways. This requires violating the condition CLmet + bile > > CLsef, the condition necessary for uptake clearance to be the RDS in the hepatic clearance of drug. To illustrate this effect, for three theoretical victim drugs, where CLmet + bile > > CLsef (CLmet + bile = 1, 10, 100 liters/min, CLsef = 0.1 liter/min, and CLsin = 1Qh), the systemic AUC ratio (AUCR) of the victim drug in the absence and presence of 10%–99% inhibition of CLmet + bile was simulated. In accordance with Food and Drug Administration guidelines, an AUCR of 1.25 was considered significant.
To illustrate that the CLmet + bile/CLsef ratio, and not the absolute magnitude of CLmet + bile and/or CLsef, determines when the RDSuptake switches to RDSall, we conducted the following simulations: the systemic AUC of the drug was simulated for CLsef values ranging from 0.1 to 10 liters/min (representing 0.1× to 10 × Qh) with CLmet + bile set to 1- to 20-fold the value of the corresponding CLsef. The simulated systemic AUCs, when the CLmet + bile/CLsef ratio was held constant, were compared with the simulated systemic AUCs when CLmet + bile/CLsef ratio varied.
Next, we defined the tipping point as the CLmet + bile/CLsef ratio, at which RDSuptake switches to RDSall. Following the same strategy, we simulated the AUCR for various CLmet + bile/CLsef ratios for victim drugs that originally had RDSuptake to illustrate the CLmet + bile/CLsef ratio at which AUCR = 1.25, thus signifying that RDSuptake switched to RDSall. The systemic AUC where the RDS is uptake was simulated such that the CLmet + bile/CLsef ratio = 1000 (AUCratio = control, CLmet + bile = 100 liters/min, CLsef = 0.1 liter/min). Then, systemic AUC was simulated for CLmet + bile/CLsef “test” ratios ranging from 0.1 to 10 (CLmet + bile = 0.01–1 liter/min, CLsef = 0.1 liters/min), and the resulting AUC (AUCratio = test) was normalized to the control simulation (AUCR = AUCratio = test/AUCratio = control). The decrease in CLmet + bile/CLsef ratio is akin to inhibition of CLmet + bile since CLsef is held constant. The CLmet + bile/CLsef ratio, which resulted in a significant change to the systemic AUC (AUCR = 1.25) compared with control, was identified as the tipping point.
To illustrate that the magnitude of CLsin contributes to the tipping point, we simulated the tipping point for CLsin values ranging from 0.01 × Qh to 4 × Qh (henceforth, for simplicity, CLsin notation will be used instead of fubCLsin since fub = 1). The tipping point can be explicitly derived from the ECM (eq. 1) by defining the RDS switch for any chosen AUCR as AUCR = RDSuptake/RDSall and solving for the CLmet + bile/CLsef ratio (eq. 2). This relationship (eq. 2 with AUCR = 1.25) was used later to identify DDI liabilities when considering CLsin magnitude and CLmet + bile/CLsef ratio of a drug:
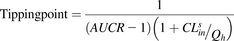 (2)
(2)Quantifying When a Drug with RDSuptake Will Switch to RDSall from Metabolic/Biliary Efflux DDIs.
We defined the PImet + bile as the percent inhibition of CLmet + bile required for RDSuptake to switch to RDSall. This quantifies when a significant DDI (AUCR ≥ 1.25) occurs from inhibition of CLmet + bile, even when uptake is the RDS in the absence of DDI. For CLmet + bile/CLsef ratios ranging from 1 to 100, CLmet + bile was inhibited 10%–99%. Simulations were conducted for CLsin values = 0.25, 1×, 4 × Qh. CLsin values were chosen to represent ER = 0.2, 0.5, and 0.8 [low-, mid-, and high-extraction ratio (ER), respectively] and were back-calculated from eq. 3 to eq. 4. The percent inhibition of CLmet + bile at which the CLmet + bile/CLsef ratio reaches the tipping point (i.e., PImet + bile) and thus causes the RDSuptake to switch to RDSall was calculated as shown in eq. 5:
 (3)
(3) (4)
(4) (5)
(5)Applying the RDS Framework to In Vitro and In Vivo Examples.
Published data sets in which all hepatobiliary clearance pathways (CLsin, CLsef, CLbile, CLmet) were quantified in vitro were collected. The in vivo hepatobiliary clearances must be used to identify the RDS of a drug. As such, the provided in vitro to in vivo extrapolated (IVIVE) clearances were used; otherwise, in vitro hepatobiliary clearance values were scaled to in vivo using IVIVE scaling factors as provided by the authors. For all drugs, fubCLsin/Qh was used to calculate the tipping point using eq. 2 (see Results section to follow). RDS was labeled as RDSuptake and RDSall if the CLmet + bile/CLsef ratio was above and below the tipping point, respectively. For drugs with RDSuptake, the PImet + bile was calculated using eq. 5. Finally, for selected drugs, the predicted DDI liabilities using the RDS and PImet+bile were compared with the observed in vivo data. To ensure that only the systemic clearance, and not the bioavailability of the victim drug, was affected, clinical DDI studies were included if the victim was a dual- transporter/enzyme substrate and coadministered with a selective enzyme inhibitor administered i.v. It should be noted that the availability of such studies was limited.
Results
Identifying the Tipping Point (i.e., When RDSuptake Switches to RDSall) and Factors that Influence this Switch.
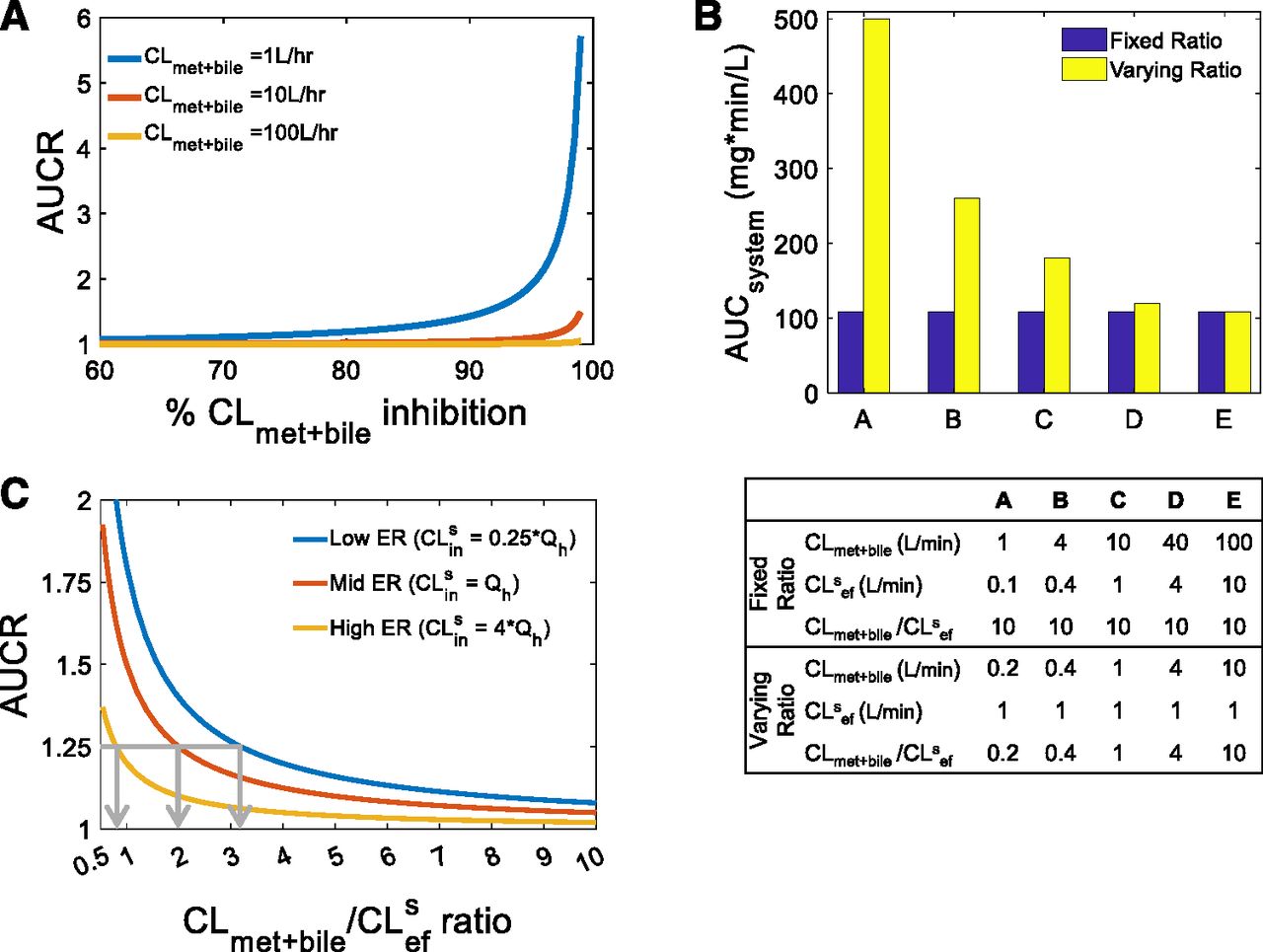
As described in the Theoretical Background section, RDSuptake occurs when CLmet + bile > > CLsef, and, as such, inhibition of CLmet + bile will not manifest in the systemic AUC of a victim drug. When this condition is violated owing to extensive inhibition of CLmet + bile, there will be a significant increase in the systemic AUC of the victim drug when CLmet + bile is inhibited further. In other words, when CLmet + bile is no longer > > CLsef, then RDSuptake switches to RDSall. In Fig. 1A, 84%, 98%, and 99.8% inhibition of CLmet + bile led to a clinically significant increase in the systemic AUC of the three theoretical victim drugs shown (AUCR ≥ 1.25). Even though the victim drugs had different preinhibition CLmet + bile values (1, 10, 100 liters/min), the postinhibition CLmet + bile values were all the same (0.2 liters/min). Since CLsef was kept constant (0.1 liters/min), an AUCR of 1.25 was observed when CLmet + bile/CLsef = 2 for all three victim drugs. This simulation illustrates that the RDSuptake switch to RDSall depends on the CLmet + bile/CLsef ratio, not on the extent of CLmet + bile inhibition.
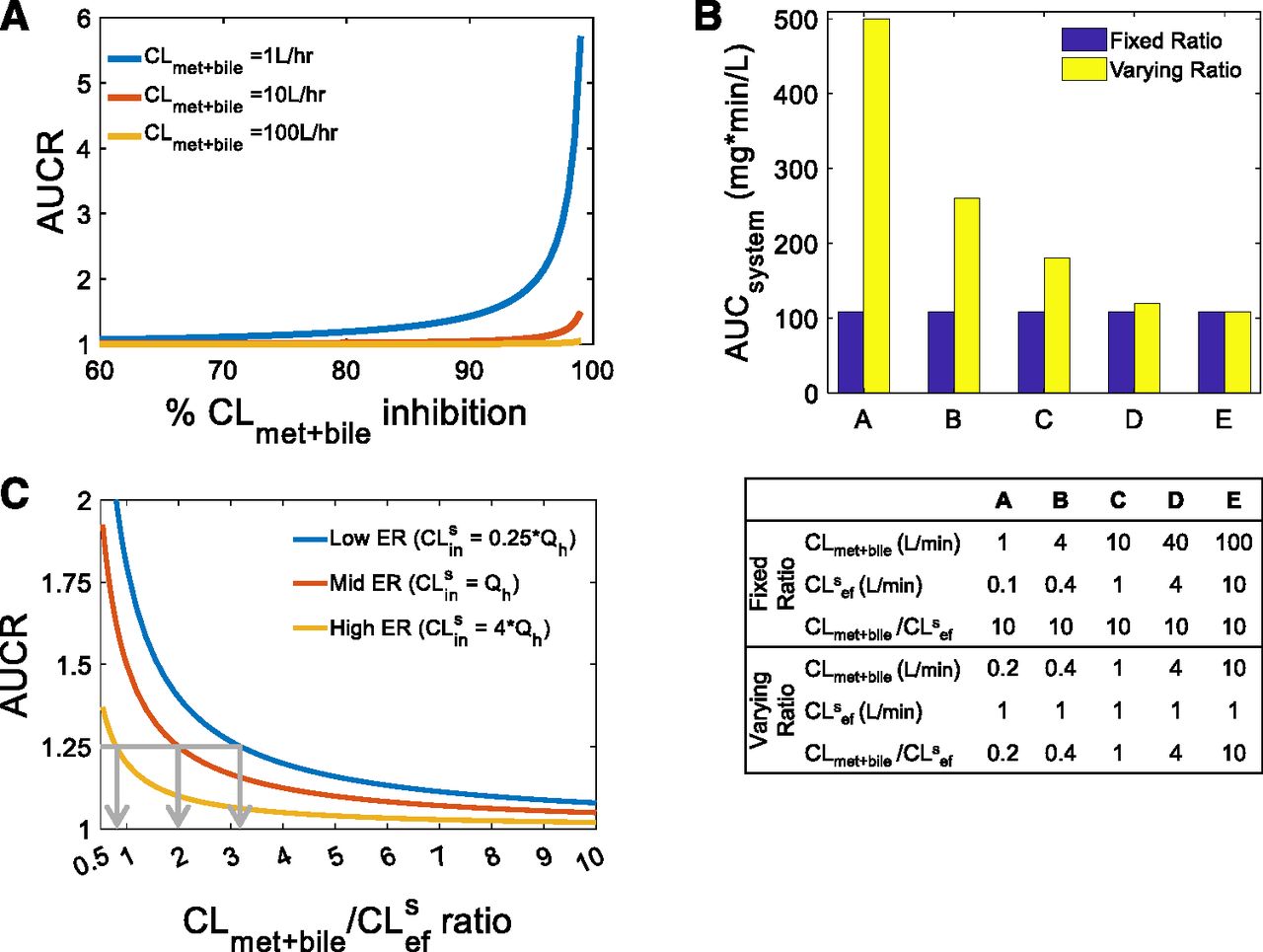
Identifying when RDSuptake switches to RDSall, that is, the tipping point. (A) Extensive inhibition of CLmet + bile can lead to a significant increase in the systemic AUC for three theoretical victim drugs that have RDSuptake (i.e., CLmet + bile > > CLsef) in the absence of DDI. When inhibition of CLmet + bile eventually violates the condition required for RDSuptake, the RDSuptake switches to RDSall. An AUCR ≥1.25 was observed when CLmet + bile was inhibited ≥84%, ≥98%, and ≥99.8% for CLmet + bile = 1, 10, 100 liters/h, respectively. For all three victim drugs, however, the CLmet + bile value after such inhibition was similar (0.2 liters/min), as was the CLmet + bile/CLsef ratio (= 2). Simulations were performed as follows: CLsin = 1xQh, CLmet + bile = 1, 10, 100 liters/h, CLsef = 0.1 liters/min. (B) The systemic AUC (in the absence of any DDI) of a theoretical drug remains unchanged when the CLmet + bile/CLsef ratio remains fixed (blue bars) but not when the CLmet + bile/CLsef ratio is varied (yellow bars), even though the absolute value of CLmet + bile and CLsef is varied in both scenarios. This trend was observed irrespective of the value of CLsin (Supplementary Fig. 1). Furthermore, this trend is true for when CLmet + bile > CLsef or CLmet + bile < CLsef (also refer to Supplementary Fig. 1). Thus, the CLmet + bile/CLsef ratio, irrespective of the magnitude of the absolute values of these clearances, is important for establishing the RDS and henceforth when the RDS switches from uptake to all hepatobiliary clearances. Simulations were performed as follows: CLsin = 0.25×Qh, and the other input clearance values for scenarios A–E are shown in the table provided. (C) Since the RDS depends on the CLmet + bile/CLsef ratio, we define the tipping point as the CLmet + bile/CLsef ratio at which RDSuptake switches to RDSall. Similarly to (A), the RDSuptake switch to RDSall is represented by an AUCR of 1.25 and the decrease in CLmet + bile/CLsef ratio is akin to inhibition of CLmet + bile when CLsef is kept constant. As shown by the gray arrows, the tipping point for a low-, mid-, and high-ER drug is 3.2, 2, and 0.8, respectively. For example, if the CLmet + bile/CLsef ratio for the low ER drug is above the tipping point (i.e., CLmet + bile/CLsef > 3.2), then the drug will have RDSuptake; therefore, DDIs from CLsin but not CLmet + bile should be expected; however, inhibition of CLmet + bile that makes the CLmet + bile/CLsef ratio lower than the tipping point (i.e., CLmet + bile/CLsef < 3.2) will lead to a significant increase in the systemic AUC. Crossing the tipping point is indicative of the RDSuptake switch to RDSall. Simulations were performed as follows: systemic AUC were simulated for CLsin = 0.25×, 1×, 4 × Qh (representing low, mid, and high ER, respectively) and CLmet + bile/CLsef ratios from 1 to 10 and then normalized to a control simulation where the CLmet + bile/CLsef ratio was set to 1000 (i.e., RDSuptake).
To further emphasize the dependence on the CLmet + bile/CLsef ratio, we simulated the systemic AUC of the victim drug (in the absence of DDI) for different CLmet + bile and CLsef values while holding CLsin constant. The systemic AUC remained unchanged when the CLmet + bile/CLsef ratio remained fixed, even though the CLmet + bile and CLsef values varied, demonstrating that the RDS in the hepatic clearance of a drug is dependent on the CLmet + bile/CLsef ratio, not on the absolute value of these clearances (Fig. 1B). This was true for both when CLmet + bile was higher and lower than CLsef (also see Supplementary Fig. 1). Since the systemic AUC decreased as the CLmet + bile/CLsef ratio increased, only the CLmet + bile/CLsef ratio needs to be considered when determining when the RDSuptake switches to RDSall for a victim drug.
Next, we identified the tipping point, defined here as the CLmet + bile/CLsef ratio when RDSuptake switches to RDSall. The RDSuptake switch to RDSall signifies when DDIs owing to inhibition of CLmet + bile start to become significant for a victim drug that has RDSuptake. As demonstrated already, the RDSuptake switch to RDSall depends on the CLmet + bile/CLsef ratio. As such, we identified the tipping point as the CLmet + bile/CLsef ratio at which the systemic AUC increases significantly (AUCR = 1.25) owing to a decrease in the CLmet + bile/CLsef ratio for a victim drug that has RDSuptake (Fig. 1C). As demonstrated in Fig. 1C, the tipping point for a low, mid, and high ER drug was 3.2, 2, and 0.8, respectively.
Since the tipping point varied for a low, mid, and high ER, the magnitude of CLsin is also an important factor in determining when the RDSuptake switches to RDSall (Fig. 1C). Extending the simulations to identify the tipping point across a range of CLsin values, we established a theoretical (eq. 2) and practical (Fig. 2) relationship between CLsin/Qh and the tipping point. The tipping point decreases as CLsin increases. In other words, as a drug’s CLsin (and therefore its ER) increases, the drug is more likely to have RDSuptake and a larger PImet + bile, therefore making the drug more resistant to switching its RDS. In addition, as the influx across the sinusoidal membrane becomes large, hepatic clearance becomes limited by blood flow and therefore less likely to result in a change in AUCR when either CLsin (or for that matter CLmet + bile) is inhibited. On the other hand, when CLsin (or ER) is small and the hepatic clearance becomes proportional to CLsin, the victim drug becomes more susceptible to a change in RDS. This demonstrates that low ER drugs are more susceptible to RDSuptake switching to RDSall, whereas high ER drugs are more resistant to the RDS switch.

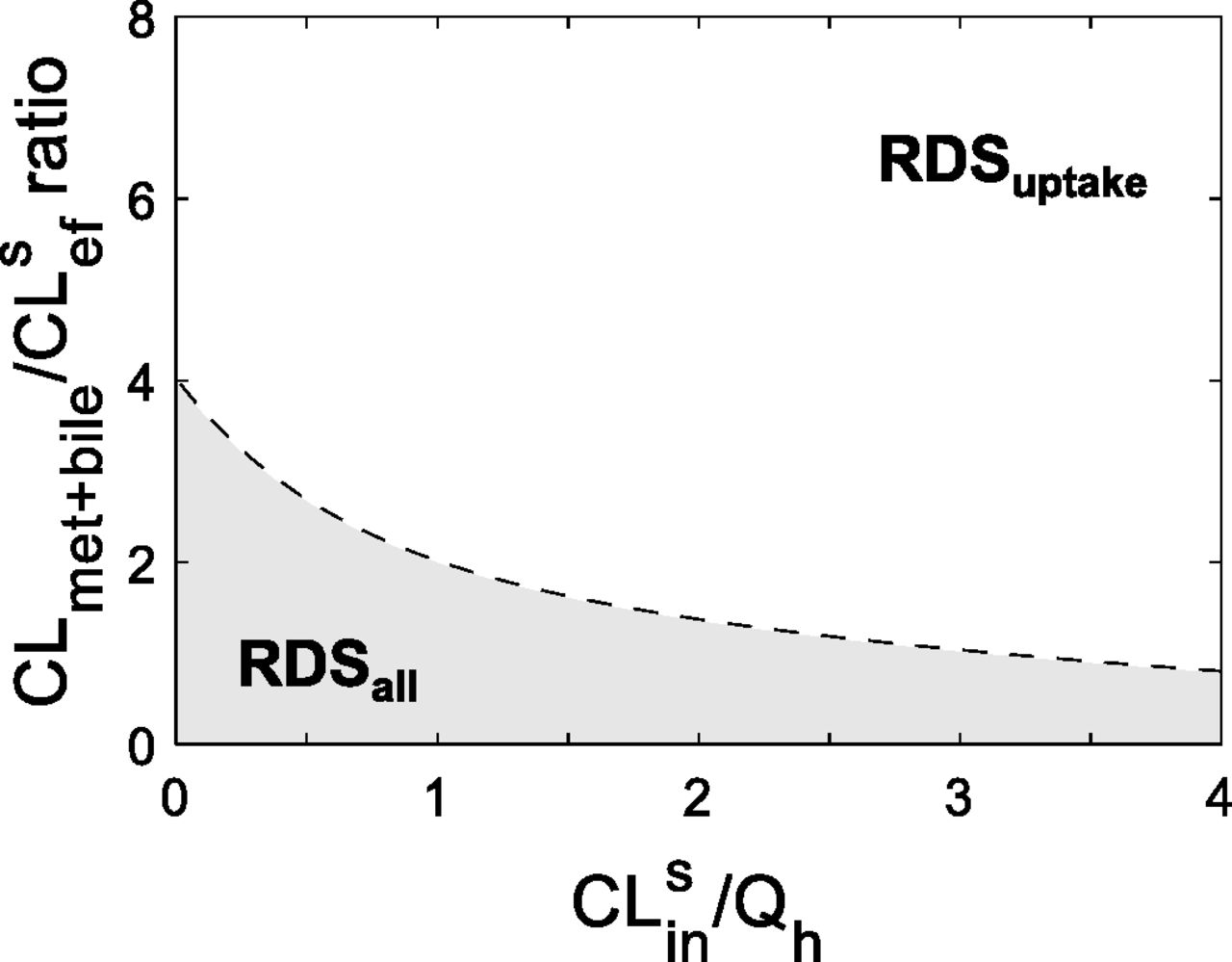
The RDS framework helps identify DDI liabilities. The CLmet + bile/CLsef ratio and CLsin magnitude of a drug determine the RDS of the drug and when RDSuptake switches to RDSall. Combinations of hepatobiliary clearances found in the shaded area have RDSall, whereas those in the nonshaded area have RDSuptake. Any alterations in hepatobiliary clearances that cause a drug to switch from the nonshaded to the shaded area will cross the tipping point (dashed line, eq. 2) and therefore switch the RDS from uptake to all hepatobiliary clearances. The consequence of this switch is that DDIs from inhibition of CLmet + bile will now manifest in the systemic AUC of a victim drug that originally had RDSuptake. Consistent with Fig. 1C, the tipping point decreases as the magnitude of CLsin (and therefore the drug’s ER) increases. This suggests that the greater the ER of the drug, the more likely it will have RDSuptake and will be more resistant to switch to RDSall. Furthermore, when CLmet + bile/CLsef > 4, the RDS will always be uptake clearance, irrespective of the value of CLsin/Qh; however, when CLmet + bile/CLsef < 4, the RDS can be either uptake or all hepatobiliary pathways, depending on the magnitude of CLsin. It should be noted that if a drug is administered orally, the tipping point will always be 4 because the blood flow limitations are no longer relevant. Simulations were performed as follows: the tipping point was simulated for CLsin values (0.01 × Qh – 4 × Qh) using eq. 2.
It should be noted that the relationship between CLsin/Qh and the tipping point (eq. 2 and Fig. 2) depends on the chosen AUCR cutoff. Here, an AUCR of 1.25 was chosen based on Food and Drug Administration guidelines of what constitutes a positive DDI. If a higher AUCR cutoff were to be selected (Supplementary Fig. 2), this would lead to estimation of lower tipping points, thus making it more likely that drugs are labeled with RDSuptake. Labeling a drug with RDSuptake when in fact it has RDSall can lead to underpredictions of DDI liabilities from metabolic enzymes and biliary transporters.
By understanding the relationship between CLsin and the tipping point, the RDS can be identified for any combination of a drug’s hepatobiliary clearance values (Fig. 2). For example, a high-ER drug with a CLmet + bile/CLsef ratio of 3 will have RDSuptake, but a low-ER drug with the same CLmet + bile/CLsef ratio will have RDSall. Furthermore, a drug will always have RDSuptake if the CLmet + bile/CLsef ratio is greater than 4, irrespective of the magnitude of CLsin. It should be noted that for orally administered drugs, the tipping point will no longer depend on the magnitude of CLsin and therefore will always be 4 because blood flow limitations from systemic clearance are cancelled out by blood flow limitations of hepatic bioavailability.
Quantifying the PImet + bile for Drugs with RDSuptake.
Identifying the RDS of a drug and when the RDSuptake to RDSall switch will happen identifies the drug’s DDI liabilities. We quantified the PImet + bile, defined here as the percent inhibition of CLmet + bile needed to cause the RDSuptake switch to RDSall, to understand when inhibition of CLmet + bile starts to become a DDI liability for victim drugs that have RDSuptake. As the CLmet + bile/CLsef ratio of the victim drug (before inhibition) increases, the PImet + bile increases (Fig. 3A) because, as CLmet + bile becomes > > than CLsef, the victim drug become resistant to the RDSuptake switch to RDSall. High-ER drugs have a higher PImet + bile than low-ER drugs, demonstrating again that high-ER drugs are resistant to the RDS switch, whereas low-ER drugs are sensitive (Fig. 3A). Figure 3B illustrates that whereas a low-, mid-, and high-ER victim drug with CLmet + bile/CLsef ratio of 6 have RDSuptake (before inhibition), inhibition of CLmet + bile >46%, 66%, and 87%, respectively, will cause the RDSuptake to switch to RDSall. This translates to observing a positive DDI owing to inhibition of CLmet + bile for a victim drug that has been identified to have RDSuptake (before inhibition). Without knowledge of the PImet + bile, such a DDI may not be expected.

Identifying when drugs with RDSuptake will start to experience a DDI from inhibition of CLmet + bile. (A) The PImet + bile, defined as the % inhibition of CLmet + bile required for the RDSuptake switch to RDSall, depends on the CLmet + bile/CLsef ratio (before inhibition) and the magnitude of CLsin (represented as low-, mid-, and high-ER drugs). The PImet + bile identifies when a positive DDI from inhibition of CLmet + bile for a drug with RDSuptake would be expected. Lower CLmet + bile/CLsef ratios, as well as low ER drugs, are the most susceptible for the RDSuptake switch to RDSall owing to CLmet + bile inhibition. (B) For RDSuptake to switch to RDSall for a theoretical victim drug with CLmet + bile/CLsef ratio of 6, CLmet + bile must be inhibited by >46%, >66%, or >87% if the drug is low-, mid-, and high-ER, respectively. Visually, the RDSuptake switch to RDSall happens when the theoretical victim drug crosses the dashed line (the tipping point) from the unshaded area (RDSuptake) to the shaded area (RDSall). Additional examples of PImet + bile are given in the table provided. Simulations were performed as follows: PImet + bile was calculated using eq. 5 for CLmet + bile/CLsef ratios ranging from 1 to 40 and for CLsin = 0.25×, 1×, 4 × Qh (representing low-, mid-, and high-ER, respectively).
The purpose and conclusions of the simulations that have been used to establish the RDS framework up to this point are summarized in Fig. 4. As discussed, identifying the drug’s RDS is not enough to predict correctly the drug’s DDI liabilities. The tipping-point concept is an important consideration when identifying DDIs for victim drugs that are dual substrates of enzymes and transporters.
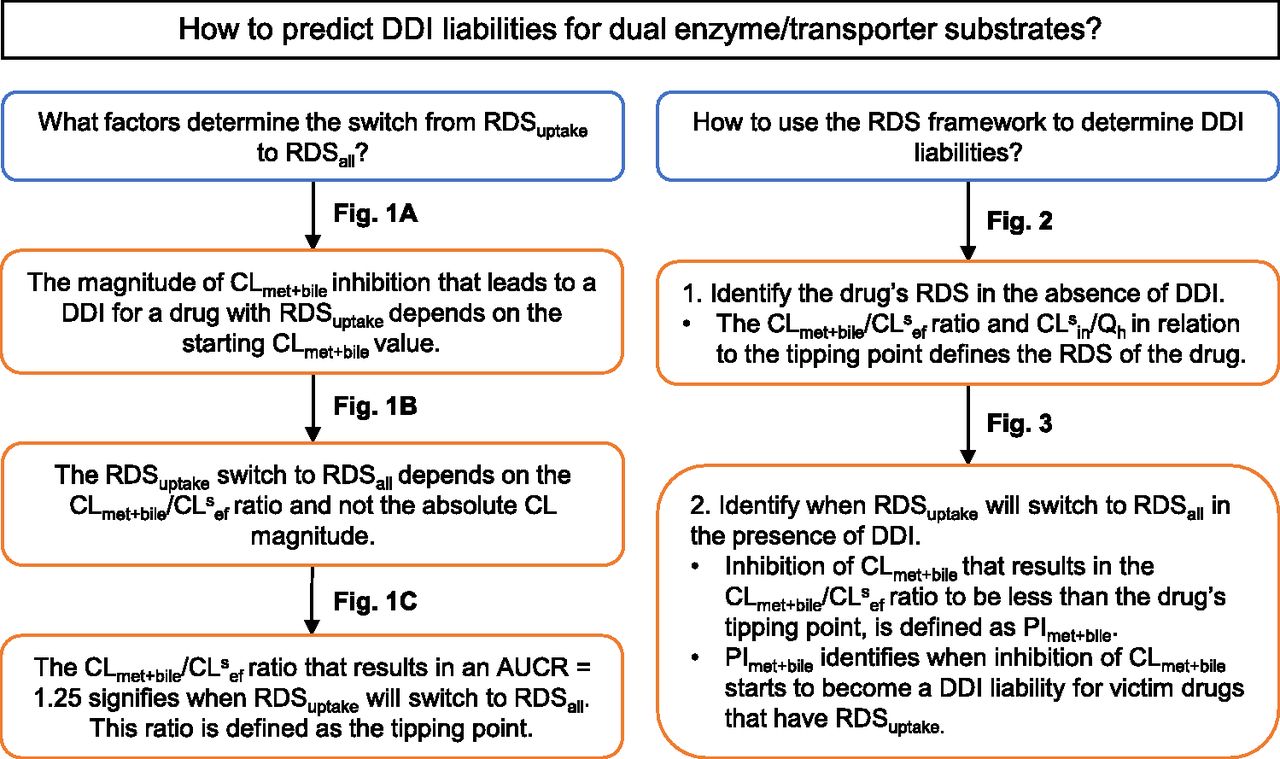
Summary of the purpose and conclusions for the simulations used to establish the RDS framework.
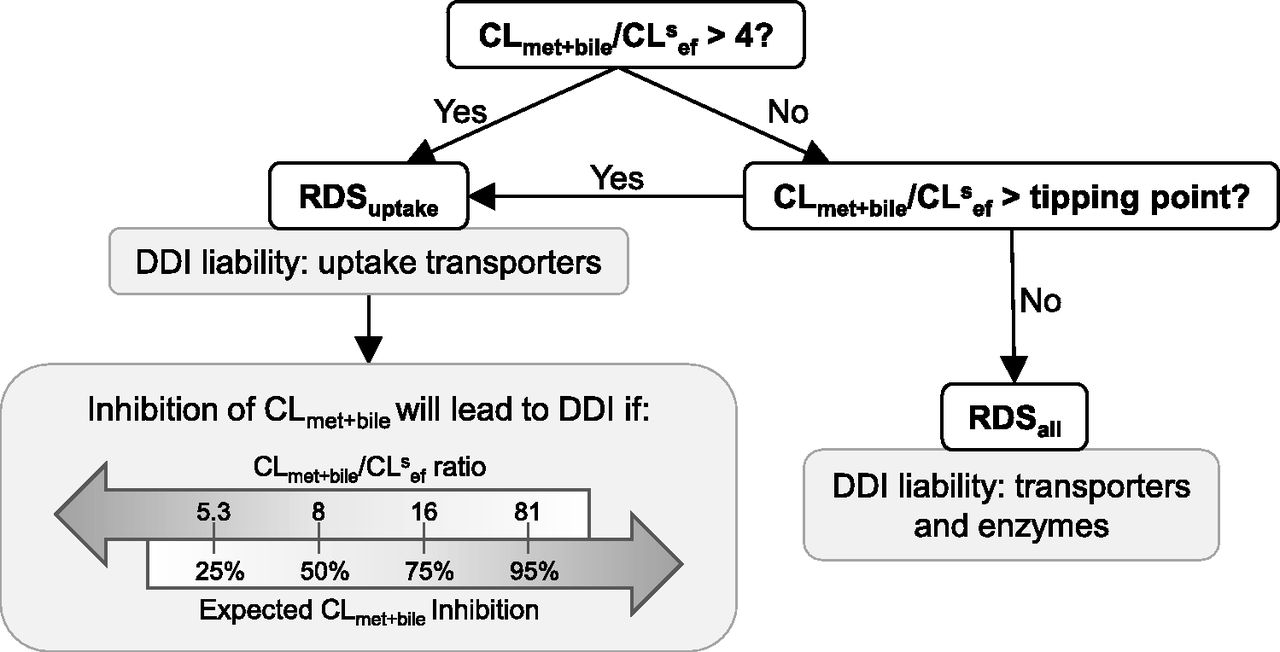
The flowchart in Fig. 5 can be used as a guide to identify the DDI liabilities for dual-transporter/enzyme substrates. All drugs with CLmet + bile/CLsef ratio >4 will have RDSuptake, whereas drugs with CLmet + bile/CLsef ratio < 4 will have RDSuptake so long as this ratio is greater than the tipping point. Drugs with CLmet + bile/CLsef ratio less than the tipping point will have RDSall. If the drug has RDSuptake, then uptake transporters will become a DDI liability, whereas if the drug has RDSall, then transporters and enzymes will be a DDI liability. Even for drugs that have RDSuptake, however, CLmet + bile can become a DDI liability if inhibition of CLmet + bile is greater than the predicted PImet + bile and thus causes the RDSuptake switch to RDSall. The flowchart identifies the CLmet + bile/CLsef ratios at which 25%, 50%, 75%, and 95% expected inhibition of CLmet + bile is going to result in the RDS switch. This information can be used to assess when CLmet + bile starts to become a DDI liability for drugs with RDSuptake. It also helps answer the question of how much larger CLmet + bile needs to be compared with CLsef for sinusoidal uptake clearance to become (and maintain) the RDS in the hepatic clearance of any drug. Such information may be used during drug development to select drug candidates if a certain RDS is desired.

Applying the RDS framework to identify DDI liabilities for dual transporter-enzyme substrate drugs. If CLmet + bile/CLsef > 4, then the drug will have RDSuptake, irrespective of the magnitude of CLsin. For drugs with RDSuptake, DDIs due to inhibition of CLmet + bile can become significant, depending on the drug’s CLmet + bile/CLsef ratio and the expected inhibition of CLmet + bile. For example, 50% inhibition of CLmet + bile may result in a significant DDI for a drug with RDSuptake and CLmet + bile/CLsef ratio <8 but no DDI will be observed if the drug has CLmet + bile/CLsef ratio >8. The DDI liability owing to inhibition of CLmet + bile increases as the CLmet + bile/CLsef ratio decrease and the expected CLmet + bile inhibition increases.
Applying the RDS Framework to In Vitro and In Vivo Examples.
To provide context to the theoretical framework presented, examples from literature, where available, were used. For drugs with in vitro–quantified hepatobiliary clearances that were extrapolated to in vivo via IVIVE, the tipping point and the PImet + bile were calculated using eq. 2 and eq. 5, and a subset of the analyzed data set, which includes primarily statin drugs, is shown in Fig. 6 (also see Supplementary Table 1) (Camenisch and Umehara, 2012; Jones et al., 2012; Varma et al., 2014; Kunze et al., 2015; Riede et al., 2017). If no empirical scaling factors (such as for active uptake clearance to match observed in vivo clearance) are included in the IVIVE process, then almost all drugs have RDSall, except valsartan and pravastatin (Fig. 6A). This is because most CLmet + bile/CLsef ratios are <4, and because the IVIVE CLsin magnitudes were small, most CLmet + bile/CLsef ratios were less than the tipping point. Because many statins have been identified to have RDSuptake, the trend in Fig. 6A suggests that CLsin was underestimated in vitro. When hepatobiliary clearances were adjusted by empirical scaling factors (Varma et al., 2014) or parameters were fitted from in vivo intravenous concentration-time profiles using a physiologically based pharmacokinetic (PBPK) model (Jones et al., 2012), the distribution of drugs is altered as low ERdrugs (ER < 0.2) tended to have RDSall, whereas mid- and high-ER drugs (ER > 0.2) were more likely to have RDSuptake (Fig. 6B). This analysis of the published in vitro hepatobiliary clearances provides insight that drugs with RDSuptake exist within the moderate RDS framework space, meaning that in general their CLmet + bile/CLsef ratio is <4, and they are quite susceptible to the RDS switch (Supplementary Table 1). It further elucidates that current in vitro quantification techniques may underestimate CLsin, which can lead to erroneous labeling of the RDS and thus incorrect DDI liability predictions (Fig. 7; Supplementary Fig. 3).

The distribution of drugs within the RDS framework using hepatobiliary clearance quantified in vitro and extrapolated to in vivo. Published in vitro hepatobiliary clearance values, when extrapolated to in vivo via IVIVE, can identify the RDS based on fubCLsin/Qh and CLmet + bile/CLsef ratio (eq. 2). (A) When no empirical scaling factors, such as to scale up active transport, are applied during the IVIVE process, all drugs except for valsartan and pravastatin have RDSall. Most drugs had CLmet + bile/CLsef ratio <4, indicating drugs primarily exist within the moderate RDS framework space. Furthermore, most drugs have fubCLsin/Qh < 0.4, indicating severe underprediction of CLsin. (B) When empirical scaling factors are used or hepatobiliary clearances are estimates from in vivo data using PBPK modeling, the RDS of the drugs is altered severely. Now, RDSuptake occurs more often for mid- and high-ER drugs with RDSall primarily for low ER drugs (ER was calculated from in vivo hepatic clearance and blood flow). Furthermore, since all drugs have CLmet + bile/CLsef ratio <4, information about both the magnitude of fubCLsin and the CLmet + bile/CLsef ratio is necessary to correctly predict DDI liabilities. The dashed line represents the tipping point (eq. 2). The data shown are from Jones et al. (2012) and Varma et al. (2014) and represent a subset of the complete data set presented in Supplementary Table 1.

The impact of underpredictions of hepatobiliary clearance on DDI liability predictions. A representative 3-fold underprediction of either (A) CLsin or (B) CLmet + bile can lead to erroneous labeling of the RDS for low-, mid-, and high-ER drugs (shown by the filled circles crossing from the nonshaded to shaded area (i.e., RDSuptake switches to RDSall). Mislabeling the RDS impacts the expected DDI risk from transporters versus enzymes. Furthermore, underpredictions of either CLsin or CLmet + bile leads to identifying both transporters and enzymes as DDI liabilities when truly only uptake transporters are the true DDI liability. Please refer to Supplementary Fig. 4 for more detailed simulations.
To illustrate more fully the applicability of the RDS framework, predicted DDI liabilities using the RDS framework were compared with in vivo DDI examples. As indicated in Table 1, when empirical scaling factors are used during the IVIVE process or hepatobiliary clearances were estimated from in vivo via PBPK, atorvastatin and repaglinide have RDSuptake and PImet + bile of 10%–51% and 15%–40%, respectively, whereas bosentan has RDSall. For atorvastatin and repaglinide, the in vitro data predicted that uptake transporters (OATPs) are the primary DDI liability, with the drugs’ major metabolic enzymes (CYP3A and CYP2C8, respectively) becoming a potential liability only if the in vivo hepatic metabolic inhibition is greater than the PImet + bile. For bosentan, the in vitro data predicted that both OATPs and CYP3A4 are potential DDI liabilities. Clinically, for atorvastatin, coadministration of rifampin (an OATP inhibitor) leads to an AUCR of 12, whereas 33% inhibition of CYP3A4 due to intravenous itraconazole (as measured using CYP3A4 probe midazolam) did not change atorvastatin systemic AUC, even though inhibition of atorvastatin metabolism was observed via a decrease in the 2-hydroxyatorvastatin concentrations (Maeda et al., 2011). In a similarly conducted experiment, coadministration of rifampin resulted in AUCR of 3.2 and 1.9 for bosentan or repaglinide, respectively, whereas 73% inhibition of CYP3A4 owing to intravenous itraconazole did not significantly change the systemic AUC of these drugs (Yoshikado et al., 2017). Furthermore, repaglinide coadministered with oral rifampin and trimethoprim (CYP2C8-selective inhibitor) resulted in AUCR 2.6 and 1.8, respectively (Kim et al., 2016). The in vivo DDI liability for OATPs was well predicted for all three victim drugs. The in vivo DDI liability for CYP3A4 was well predicted for atorvastatin. Since a probe was not used to assess the degree of CYP2C8 inhibition, it is difficult to determine whether the significant DDI when repaglinide was coadministered with trimethoprim was because RDSuptake switched to RDSall or because repaglinide truly has RDSall. The in vitro metrics, as well as a whole-body PBPK DDI model, suggests that repaglinide has RDSuptake (Varma et al., 2013); thus, the repaglinide-trimethoprim DDI is likely due to the RDS switch. Lastly, since bosentan was predicted to have RDSall, a DDI was expected to result from CYP3A4 inhibition, but none was observed. It should be noted that the metabolic DDI liability prediction is assuming one main drug-metabolizing enzyme and no significant biliary efflux (e.g., CLmet + bile = CLCYP3A4 for atorvastatin and bosentan). This assumption predicts the highest DDI risk owing to inhibition of CLmet + bile and has a higher chance of predicting false-positive DDI results.
Comparison of predicted drug-drug interaction (DDI) liabilities from in vitro data to in vivo clinical studies
Hepatobiliary clearances, after in vitro to in vivo extrapolation (IVIVE), can be used to identify the rate-determining step (RDS) of a drug, such as if the CLmet + bile/CLsef ratio is > or < than the tipping point (eq. 2), then the drug will have RDSuptake or RDSall, respectively. If a drug has RDSuptake, then the PImet + bile can be quantified (eq. 5) to predict when a significant DDI should be expected owing to inhibition of metabolic/biliary efflux clearance. An expanded analysis is shown in Supplementary Table 1.
In the published in vitro data sets, discrepancies in the in vitro quantified values, particularly for CLsin, can be observed (Supplementary Table 1; Table 1). For example, in one report, the authors used empirical scaling factors for active sinusoidal uptake clearance to match hepatic clearance with clinically observed data that ranged from 1.1 to 101.8 with a geometric mean of 10.6 (Varma et al., 2014); however, the scaling factor used severely impacted the labeling of the RDS (e.g., fluvastatin, glyburide, pravastatin) or impacted the predicted PImet + bile of drugs (e.g., atorvastatin, rosuvastatin, fluvastatin, repaglinide) (Supplementary Table 1). Assumptions regarding CLsef also caused discrepancies. In all reports, CLsef was assumed to be equal to passive diffusion across the sinusoidal membrane, except in one report in which CLsef was back-calculated from total sandwich cultured human hepatocytes CLint (Camenisch and Umehara, 2012). The assumptions surrounding CLsef impacted the CLmet + bile/CLsef ratio, which either changed how the RDS was labeled or the magnitude of the PImet + bile (e.g., aliskerin, ciprofloxacin, digoxin) (Supplementary Table 1). All in all, mispredictions of any of the hepatobiliary clearances impact the RDS labeling, magnitude of the PImet + bile, and DDI liability predictions.
Errors from in vitro quantification of hepatobiliary clearances can propagate when establishing the RDS and the predicted DDI liabilities. Underprediction of both CLsin and CLmet + bile may erroneously label a drug with RDSall when it is truly RDSuptake (Fig. 7). CLmet + bile is the more sensitive parameter for determining the RDS because underpredictions of CLsin may mislabel the RDS only for drugs with CLmet + bile/CLsef ratio < 4 (Supplementary Fig. 4). For such drugs, even moderate (e.g., 2- to 5-fold) underpredictions of either clearance pathway will lead to RDS mislabeling (Supplementary Fig. 4). Furthermore, underpredictions of both CLsin and CLmet + bile leads to underprediction of PImet + bile, resulting in predicting a larger DDI liability owing to CLmet + bile inhibition for a drug with RDSuptake (Fig. 7; Supplementary Fig. 4). Whereas underpredictions of hepatobiliary clearances will result in conservative DDI decisions, they also increase the chances of negative DDI studies.
Discussion
We built a theoretical RDS framework and identified important considerations when predicting DDI liabilities for dual transporter-enzyme substrate drugs. First, inhibition of CLmet + bile can cause the RDS of a victim drug to switch from RDSuptake to RDSall and hence result in an unexpected systemic DDI. Two metrics have been developed to identify when the RDS switch occurs: the tipping point, defined as the CLmet + bile/CLsef ratio at which RDSuptake will switch to RDSall, and the PImet + bile, defined as the percent inhibition of CLmet + bile at which a significant AUC change (AUCR >1.25) for a drug with RDSuptake will start to be observed. The tipping point depends on the drug’s CLmet + bile/CLsef ratio and on the magnitude of CLsin. The former but not the latter condition is relevant when victim drugs are administered orally. Second, we showed that the CLmet + bile/CLsef ratio must be >4 for any drug to have RDSuptake. Third, we applied the RDS framework to in vitro–quantified hepatobiliary clearances and observed that most drugs have CLmet + bile/CLsef ratio < 4; hence, in practice, the magnitude of CLsin must be considered when establishing the RDS.
Our theoretical analysis demonstrates that the CLmet + bile/CLsef ratio, and not the absolute magnitudes of the clearances, determines the RDS in the hepatic clearance of a drug. Previous publications allude to this relationship. The authors of the ECCCS observed through experimental data that when CLmet + bile is 2 × CLsef, drugs that have RDSuptake can be separated from those that do not (Riede et al., 2016). Furthermore, the β value [β = CLmet + bile/(CLmet + bile + CLsef)] introduced by Yoshikado et al. (2016) can be used to differentiate the RDS, such as when β approaches unity (i.e., CLmet + bile > > CLsef), a drug has RDSuptake. Our analyses corroborate and expand upon these results to provide a quantitative definition of the demarcation point between RDSuptake and RDSall (i.e., the tipping point) and illustrate that the magnitude of CLsin, in addition to the CLmet + bile/CLsef ratio, is an important factor in determining the RDS of a drug. That is, as a drug’s CLsin value increases, the drug is more likely to have RDSuptake and to become resistant to the RDSuptake switch to RDSall.
We found good agreement for atorvastatin in vivo to predict DDI liabilities (Table 1). For bosentan, overprediction of expected DDI owing to inhibition of CLmet + bile may be due to errors in the quantification of the hepatobiliary clearances. Indeed, a study in cynomolgus monkeys, in which bosentan plasma and liver drug concentrations were quantified, found that the in vitro scaled CLsin and CLmet were 28- and 13-fold underpredicted, whereas CLsef (assumed equal to passive diffusion) was overpredicted by 2-fold compared with the in vivo–fitted values (Morse et al., 2017). Combining the in vitro metrics that identify RDSuptake for repaglinide with in vivo repaglinide DDIs, it appears that CYP2C8 but not CYP3A4 inhibition may lead to RDSuptake switch to RDSall. Indeed, inhibition of repaglinide with gemfibrozil (CYP2C8 and OATP1B1 inhibitor) led to an 8-fold increase in systemic AUC; coadministration of itraconazole or cyclosporine (OATP1B1 and CYP3A4 inhibitor) led to much more modest 1.4- and 2.4-fold increases in systemic AUC (Niemi et al., 2003; Kajosaari et al., 2005).
The DDI liabilities discussed so far are relevant for systemic drug exposure but not necessarily for hepatic drug exposure, and thus efficacy/toxicity, if the site of action is in the liver. For example, the low-density lipoprotein cholesterol-lowering effect mediated by atorvastatin does not change for subjects with OATP1B1 polymorphism c.521T>C, even though there is a significant increase in atorvastatin systemic AUC (Maeda, 2015). This is because if the liver is the main eliminating organ, changes to sinusoidal uptake alter the hepatic concentration-time profile but not the hepatic AUC; however, a systemic increase of atorvastatin may lead to off-target toxicity, such as muscle myopathy. We refer the readers to our previous publication (Patilea-Vrana and Unadkat, 2016), which describes simulations that demonstrate the impact of inhibition of uptake or metabolism on both systemic and hepatic AUC when the liver is and is not the main eliminating organ.
The contrast between in vitro quantified CLsin with and without empirical scaling factors in Fig. 6 demonstrates that IVIVE of accurate transporter-mediated clearance remains challenging (Chu et al., 2013; Feng et al., 2014). The system used for in vitro quantification may be crucial since CLsin for statins quantified in sandwich cultured human hepatocytes appeared to be lower in magnitude than when quantified in suspended hepatocytes (Supplementary Table 1; Table 1). This may be mediated by significant intracellular localization of plasma membrane transporters (Kumar et al., 2017) or high interindividual variability when using individual donors (Vildhede et al., 2014). These reasons may also cause underpredictions of CLsin or CLbile. For transporter IVIVE, we have previously recommended using a bottom-up proteomic approach and adjusting for in vitro activity via IVIVE transporter expression–based scaling factors (Prasad and Unadkat, 2014). We have recently demonstrated the successful prediction of hepatobiliary clearance of rosuvastatin in rats using the aforementioned approach (Ishida et al., 2018).
Special emphasis needs to be given to quantifying CLsef along with CLmet + bile since the CLmet + bile/CLsef ratio is one of the anchor points when establishing the RDS. Because CLsef is a difficult parameter to quantify in vitro, it is typically assumed to be equal to passive diffusion across the sinusoidal membrane; however, there are examples of active sinusoidal efflux transport, such as MRP3 efflux of rosuvastatin (Pfeifer et al., 2013). Active sinusoidal efflux would increase the magnitude of CLsef and decrease the CLmet + bile/CLsef ratio, making a drug more likely to have RDSall. One approach to measuring CLsef is to use an integrative temporal modeling approach in sandwich cultured hepatocytes (Pfeifer et al., 2013; Ishida et al., 2018).
Errors in the quantification of CLsin and/or the CLmet + bile/CLsef ratio can impact DDI liability predictions. For example, patients with OATP1B1 polymorphism c.521T>C have about a 2-fold greater atorvastatin AUC compared with the wild-type allele (Maeda, 2015). Because of the lower CLsin, and therefore greater susceptibility to the RDSuptake to RDSall switch, patients with OATP1B1 polymorphism may experience a DDI attributable to inhibition of CYP3A, whereas patients with the wild-type allele may not. The same trend would be true for patients with polymorphic enzymes that result in lower CLmet + bile and thus lower CLmet + bile/CLsef ratios. Polypharmacy use can also impact DDI liability predictions. For example, highly active antiretroviral therapy typically includes potent CYP3A4 and moderate OATP inhibitor ritonavir, among other drugs, which can impact the CLmet + bile/CLsef ratio more severely than if only one drug is administered. Indeed, the systemic AUC of atorvastatin increased by 3.9- and 9.4-fold when coadministered with saquinavir/ritonavir and tipranavir/ritonavir, respectively (Fichtenbaum et al., 2002; Pham et al., 2009). Lastly, the saturation of enzymes, leading to a lower CLmet with increased dose, may lower the CLmet + bile/CLsef ratio and cause DDIs owing to the RDSuptake switch to RDSall.
If a victim drug has RDSall, but it has been mislabeled as RDSuptake, then the DDI liability owing to inhibition of both transporter and metabolic activity could be underestimated. Considering potential DDI risks, it would be most conservative to assume a drug has RDSall; however, making such an assumption would lead to an increase in negative DDI studies, particularly when conducting metabolic or biliary efflux DDI studies if the drug has RDSuptake. An analysis of the DDIs performed for a cohort of NMEs in 2013 showed a modest return on investment because 57% (n = 141) of all in vivo DDIs were negative (Lesko and Lagishetty, 2016). Given the high prevalence of negative DDIs, it may be more appropriate to make mechanistic-based rather than conservative decisions regarding DDI liabilities.
The RDS framework presented here should be used as a guide for identifying the DDI liabilities, whereas PBPK models should be used to predict the direction and magnitude of complex transporter-enzyme DDIs. Several examples of such models (e.g., repaglinide, simvastatin, rosuvastatin) exist that predict complex interactions resulting from chemical inhibition or genetic polymorphism (Varma et al., 2013; Rose et al., 2014; Tsamandouras et al., 2015). Even with PBPK models, there are limitations. For example, when a drug has RDSuptake, the CLmet + bile is unidentifiable from plasma concentrations data since only CLsin plays a significant role in determining hepatic clearance. Focusing on capturing the correct CLmet + bile magnitude, and not the CLmet + bile/CLsef ratio, can be misleading and will impact PBPK predictions. For instance, in an atorvastatin PBPK model, when cyclosporine CYP3A4 Ki was modulated 100-fold, a maximum 1.6–fold AUCR was achieved (Duan et al., 2017). Although the tendency is to run sensitivity analysis on the active components (transport and metabolism), a sensitivity analysis on CLsef value (in the model, it was assumed to be equal to passive diffusion) should also be run as, for the specific example provided, it would likely have revealed a larger impact of cyclosporine on atorvastatin systemic AUC. Such an analysis may be helpful in consolidating in vitro Ki data with observed in vivo DDI data.
In summary, we introduced a theoretical RDS framework to predict more completely DDI liabilities for drugs that are dual transporter-enzyme substrates. We provide useful insights, such as the following: 1) the RDSuptake switch to RDSall depends on the ratio of CLmet + bile/CLsef and the magnitude of CLsin; 2) CLmet + bile/CLsef ratio > 4 ensures RDSuptake independent of CLsin magnitude or administration route; 3) there are existing drugs within a moderate space within the RDS framework that are susceptible to the RDSuptake switch to RDSall. Whereas these insights were obtained from the hepatic ECM, they can be equally applied to other organs, such as the kidneys, in which vectorial (basal-to-apical) transport of drugs is possible.
Acknowledgments
We thank Bhagwat Prasad and Neha Maharao for insightful and constructive comments and suggestions, as well as the anonymous reviewers, who gave excellent feedback to improve the clarity and significance of this work and whose suggestions prompted us to derive the explicit solution for the tipping point.
Authorship Contributions
Participated in research design: Patilea-Vrana, Unadkat.
Conducted experiments: Patilea-Vrana.
Contributed new reagents or analytical tool: Patilea-Vrana.
Performed data analysis: Patilea-Vrana.
Wrote or contributed to the writing of the manuscript: Patilea-Vrana, Unadkat.
Footnotes
- Received March 7, 2018.
- Accepted August 10, 2018.
This research was supported in part by National Institutes of Health National Institute of Drug Abuse [Grant P01DA032507].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- AUCR
- area under the curve ratio
- CLbile
- biliary (canalicular) efflux clearance
- CLint
- intrinsic clearance
- CLmet
- metabolic clearance
- CLsin
- sinusoidal influx clearance
- CLsef
- sinusoidal efflux clearance
- DDI
- drug-drug interaction
- ECM
- extended clearance model
- IVIVE
- in vitro to in vivo extrapolation
- NME
- new molecular entity
- OATP
- organic anion transporting polypeptide
- PBPK
- physiologically based pharmacokinetics
- PImet + bile
- percent inhibition of CLmet + bile necessary for RDSuptake to switch to RDSall
- RDS
- rate-determining step
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}