


Abstract
Metabolic profiles of four drugs possessing diverse metabolic pathways (timolol, meloxicam, linezolid, and XK469) were compared following incubations in both suspended cryopreserved human hepatocytes and the HμREL hepatocyte coculture model. In general, minimal metabolism was observed following 4-hour incubations in both suspended hepatocytes and the HμREL model, whereas incubations conducted up to 7 days in the HμREL coculture model resulted in more robust metabolic turnover. In the case of timolol, in vivo human data suggest that 22% of the dose is transformed via multistep oxidative opening of the morpholine moiety. Only the first-step oxidation was detected in suspended hepatocytes, whereas the relevant downstream metabolites were produced in the HµREL model. For meloxicam, both the hydroxymethyl and subsequent carboxylic acid metabolites were abundant following incubation in the HμREL model, while only a trace amount of the hydroxymethyl metabolite was observed in suspension. Similar to timolol, linezolid generated substantially higher levels of morpholine ring-opened carboxylic acid metabolites in the HμREL model. Finally, while the major aldehyde oxidase–mediated mono-oxidative metabolite of XK469 was minimally produced in hepatocyte suspension, the HμREL model robustly produced this metabolite, consistent with a pathway reported to account for 54% of the total urinary excretion in human. In addition, low-level taurine and glycine conjugates were identified in the HµREL model. In summary, continuous metabolite production was observed for up to 7 days of incubation in the HµREL model, covering cytochrome P450, aldehyde oxidase, and numerous conjugative pathways, while predominant metabolites correlated with relevant metabolites reported in human in vivo studies.
Introduction
In vitro studies directed at identifying relevant metabolites (i.e., biotransformation pathways) of drug candidates are a current expectation in drug discovery and development due to regulatory agency guidelines that focus on metabolites in safety and drug-drug interactions (USFDA, 2016, 2017). The objective of metabolite identification studies can vary depending on the stage of the program, ranging from early metabolic soft-spot assessment toward guiding medicinal chemistry strategies to improve metabolic stability to later-staged cross-species comparison of metabolic profiles to support preclinical toxicology species selection and to provide an early prediction of possible in vivo human metabolism. It is also important to identify the enzyme(s) involved in generating key metabolites (reaction phenotyping). Conventional in vitro systems for evaluating metabolism include liver subcellular fractions (e.g., S9 fraction, cytosol, or microsomes) and hepatocytes (Anderson et al., 2009; Dalvie et al., 2009), each of which have advantages and disadvantages depending on the intended purpose. One disadvantage of subcellular fractions is that they only contain a subset of drug metabolizing enzymes, depending on where they are localized. With cryopreservation techniques advancing, cryopreserved hepatocytes have become more frequently used since they contain the full complement of oxidative, hydrolytic, and conjugative enzymes, and thus provide a more comprehensive prediction of metabolism for the corresponding in vivo system (McGinnity et al., 2004; Brown et al., 2007).
In early drug discovery programs during lead optimization, one of the common parameters that drug metabolism scientists and medicinal chemists need to optimize is metabolic stability, in an effort to identify a drug candidate with likelihood of a reasonably low clearance and therapeutic dose. Metabolic soft-spot analysis in simple in vitro systems such as microsomes or hepatocytes is well suited for this objective. However, once this drug property is optimized and metabolically stable compounds become more common, another major challenge surfaces, which is the ability to robustly characterize and differentiate low-turnover (i.e., slowly metabolized) drug candidates from both a clearance rate and metabolite production standpoint. Slowly metabolized drugs require longer incubation times to enable a robust signal that can be measured with confidence (Di and Obach, 2015; Hutzler et al., 2015), which is not practical using conventional in vitro systems. For example, the general consensus is that metabolic enzyme activity in incubations employing human liver microsomes is only robust for roughly 1 hour (Foti and Fisher, 2004). In addition, hepatocytes in suspension have been reported to only maintain activity for roughly 4 hours (Di and Obach, 2015), while plating hepatocytes in a monoculture prolongs activity to approximately 24 hours, although baseline enzyme activity relative to suspension is typically less (Smith et al., 2012). In addition, 24-hour incubation times may still be insufficient for measuring turnover. As a result, novel in vitro methodologies, including the relay method using a repeat series of 4-hour incubations with suspended hepatocytes, and micropatterned coculture hepatocyte systems where incubations are conducted for up to 7 days, have been reported to perform well in the production of human relevant metabolites, both for low-turnover drugs as well as drugs that undergo multistep biotransformation reactions (Wang et al., 2010; Ballard et al., 2014).
The primary objective of this study was to evaluate the HμREL human hepatocyte coculture system as a suitable in vitro model for generation of metabolites for low-turnover drugs consistent with those observed in previously reported human clinical studies. Four commercially available drugs (timolol, meloxicam, linezolid, and XK469) were incubated for 4 hours using a conventional pooled human hepatocyte suspension and metabolic profiles were compared with those generated in the HμREL hepatocyte coculture model following incubation for up to 7 days. Findings from this appraisal of the HμREL coculture model, and the correlation with metabolic profiles in human clinical metabolism studies, are reported herein.
Materials and Methods
Chemical and Biologic Reagents.
Timolol, meloxicam, linezolid, and XK469 were purchased from Sigma Chemicals (St. Louis, MO). Pooled cryopreserved human hepatocytes (five-donor mixed gender, lot HSH), hepatocyte media (InVitroGro KHB for incubation), and additional cell culture reagents were obtained from BioreclamationIVT (Baltimore, MD). Cryopreserved hepatocyte recovery medium was obtained from APSciences, Inc. (Columbia, MD). HµREL human hepatocyte coculture plates (24-well format) were obtained from HµREL Corporation (North Brunswick, NJ). All other reagents and chemicals were of the highest purity available.
Hepatocyte Suspension Incubations.
Pooled cryopreserved human hepatocytes were stored in liquid nitrogen until use. Immediately before experiments, vials of hepatocytes were thawed rapidly (75–90 seconds) by gently shaking in a water bath at 37°C. The contents of each vial were diluted into 50 ml of prewarmed (37°C) cryopreserved hepatocyte recovery medium and gently mixed before centrifugation at 100g for 10 minutes at room temperature. The hepatocyte pellet was resuspended in InVitroGro KHB and the cell number and viability were determined using a Nexcelom Cellometer K2 Image Cytometer (Nexcelcom, Lawrence, MA). Hepatocyte viabilities were ≥93%. Cryopreserved hepatocyte suspensions were then diluted to 500,000 cells per incubation (0.5 × 106 cells/ml in 1 ml incubation) using InVitroGro KHB. Stock solutions of test compounds (50 mM in dimethylsulfoxide) were prepared and diluted to 1 mM in acetonitrile (10 µl of stock solution into 490 µl acetonitrile). A small volume (10 µl) of the new stock was added directly to 990 µl of the hepatocyte suspension to initiate the incubation, giving a final concentration of 10 µM (0.02% dimethylsulfoxide and 0.98% acetonitrile). A control sample (no cell control) was prepared by adding the same volume of test compound directly to InVitroGro KHB only. Verapamil (2 µM) was incubated as a positive control. Following 4 hours of incubation at 37°C in an atmosphere of 5% CO2 and 95% relative humidity while shaking at 400 rpm, samples were quenched with an equal volume of chilled acetonitrile. Samples were centrifuged at 4000 rpm for 10 minutes and an aliquot of each supernatant was diluted 1:4 (v/v) in solvent A (0.1% formic acid in 10 mM ammonium formate) for data-independent acquisition (DIA) analyses using high-resolution mass spectrometry (HRMS). Aliquots (600 µl) of selected supernatant were dried, reconstituted in acetonitrile (30 µl) and solvent A (30 µl), and diluted 1:4 (v/v) in solvent A for data-dependent acquisition (DDA) analyses using HRMS. Control incubation samples (verapamil) were analyzed and compared with historical data to ensure metabolic competence of the hepatocytes (data not shown).
HµREL Hepatocyte Coculture Incubations.
Cryopreserved pooled human hepatocytes (five-donor mixed gender, lot 1410235; Xenotech LLC, Kansas City, KS) and nonparenchymal stromal cells were cultured with HµREL PlatinumHeps media by HµREL Corporation. Hepatocytes, along with nonparenchymal stromal cells, were seeded into collagen-coated 24-well plates at a density of 188,000 hepatocytes per well. An additional plate containing only nonparenchymal stromal cells was also prepared. After culturing for 7 days to stabilize the cultures, plates were shipped (next day) to Q2 Solutions (Indianapolis, IN). Upon receipt, proprietary shipping media were replaced with HµREL PlatinumHeps maintenance media. After a brief acclimation period (4 hours at 37°C in an atmosphere of 5% CO2 and 95% relative humidity), HµREL PlatinumHeps maintenance media were replaced with 100 µl of HµREL PlatinumHeps dosing media containing the appropriate test compounds (final concentration of 10 µM). Plates were incubated with gentle swirling of the plates (approximately 120 rpm) at 37°C in an atmosphere of 5% CO2 and 95% relative humidity. Following 4, 24, 72, and 168 hours of incubation, supernatant from the appropriate wells was transferred to another 96-well plate. Then, 100 µl of ice cold acetonitrile was added to the incubation wells, manually mixed with the pipette to capture all cellular components, transferred, and combined with previously transferred incubation supernatant. Samples were centrifuged at 4000 rpm for 10 minutes and an aliquot of each supernatant was diluted 1:4 (v/v) in solvent A (0.1% formic acid in 10 mM ammonium formate) for DIA analyses using HRMS. Aliquots (600 µl) of selected supernatant were dried, reconstituted in acetonitrile (30 µl) and solvent A (30 µl), and diluted 1:4 (v/v) in solvent A for DDA analyses using HRMS.
Mass Spectrometry Analysis.
Metabolite profiles were generated using a high-resolution LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with an electrospray ionization source and connected to a Nexera X2 liquid chromatography system (Shimadzu Corporation, Kyoto, Japan). Chromatographic separations were performed on a Supelco Ascentis Express C18 column (2.7 µm, 2.1 × 150 mm; Sigma-Aldrich, St. Louis, MO) at 25°C using a gradient elution with solvent A (0.1% formic acid in 10 mM ammonium formate) and solvent B (acetonitrile). The gradient was initiated at a 95% A/5% B ratio at a flow rate of 0.15 ml/min, held for 2 minutes, ramped linearly from 5% B to X% B over 40 minutes (where X = 20% for samples incubated with timolol, X = 60% for samples incubated with meloxicam, X = 25% for samples incubated with linezolid, and X = 70% for samples incubated with XK469), and then ramped linearly to 95% B over the next 0.1 minute and held for 5 minutes. The column re-equilibration time was 13 minutes (total run time of 60 minutes). The mass spectrometer was operated in positive ion mode at a source voltage of 5 kV. The N2 sheath gas flow was set at 80 U, the N2 auxiliary gas flow was set at 20 U, and the capillary temperature was set at 350°C. The mass range of full-scan mass spectrometry (MS) was 100–1000 Da and the resolution was set at 30,000. The tandem MS (MS/MS) spectra for DIA were obtained using source-induced collision energy of 50 V for analyses of diluted supernatant. The MS/MS spectra for DDA were obtained using higher-energy C-trap dissociation normalized collision energies of 50, 25, 50, and 40 V, for dried, reconstituted supernatant incubated with timolol, meloxicam, linezolid, and XK469, respectively.
Metabolite Identification from Suspension and HµREL Coculture.
Accurate mass MS and MS/MS data were obtained using a Thermo Fisher LTQ-Orbitrap XL (Thermo Fisher Scientific) coupled to a Shimadzu 10AD VP HPLC (Shimadzu Corporation). The MS data were analyzed for predicted biotransformation pathways as well as metabolites previously reported in human in vivo studies. Accurate mass MS/MS data were acquired using DIA, and the MS/MS data were searched for parent drug fragment ions as well as predicted metabolite fragment ions. Metabolite target lists were generated from the MS (predicted metabolite hits) and MS/MS (DIA-predicted metabolite fragment hits) data. DDA was used to obtain more definitive metabolite structural information. Metabolite structures were correlated with literature data obtained from human clinical studies.
Results and Discussion
With low-turnover drug molecules becoming more prevalent due to successful medicinal chemistry optimization of metabolic stability properties, the challenge of how to adequately characterize and differentiate drug candidates that are slowly metabolized has come to the forefront as one of the top issues in the drug metabolism field today (Di and Obach, 2015; Hutzler et al., 2015). This challenge has forced the drug metabolism community to investigate novel assay formats that enable longer incubation times in order to allow for sufficiently measurable metabolism to occur for slowly metabolized drugs (Wang et al., 2010; Ballard et al., 2014). This work from Pfizer has been recently expanded to also include an appraisal of the multispecies micropatterned coculture system using drugs known to have species-specific metabolism (Ballard et al., 2016), a critical endeavor when selecting preclinical species for in vivo pharmacology or toxicology testing. An additional in vitro hepatocyte coculture model has also become available (HµREL), with an initial appraisal of the performance of this system for predicting intrinsic clearance and metabolite identification published by two teams at AstraZeneca (Bonn et al., 2016; Hultman et al., 2016). To further investigate the performance of the HµREL human hepatocyte coculture system, we selected four commercially available drugs (timolol, meloxicam, linezolid, and XK469) and incubated them for 4 hours using a conventional pooled human hepatocyte suspension in order to compare metabolite profiles to those generated in the HμREL hepatocyte coculture model following incubation for up to 7 days, an extension beyond previously published data where incubations were conducted for up to 3 days. In addition, the metabolites generated in vitro in both suspended hepatocytes and the HμREL human hepatocyte coculture model were compared with published human in vivo metabolism data.
Timolol.
Incubation of timolol in a suspension of cryopreserved pooled human hepatocytes for 4 hours, a commonly used in vitro methodology in drug metabolism laboratories to estimate and compare major metabolites in human and preclinical species, resulted in minimal metabolic turnover, producing only one minor mono-oxidative metabolite, T5 (Fig. 1A). In contrast, following incubation in the HμREL hepatocyte coculture model for up to 7 days, more robust metabolite generation was observed (Fig. 1A). In particular, by 24 hours of incubation, multiple oxidation steps on the morpholine ring resulted in formation of T3 and T4 as well as the terminal product, T2 (Fig. 1). It was also apparent that these multistep metabolites continued to accumulate over the 7 days of incubation (Fig. 1A). Both T2 and T3 metabolites of timolol have previously been reported to be the major urinary metabolites of timolol in man (Tocco et al., 1980). Thus, it appears that an in vitro system that allows for longer incubation time of timolol results in the generation of multistep metabolites that are consistent with the reported in vivo metabolism in human. Meanwhile, oxidation of the oxypropanolamine sidechain of timolol, which was reported as a minor pathway in vivo in human (Tocco et al., 1980), was not observed with either the conventional or HμREL hepatocyte coculture incubations (Fig. 1B). Interestingly, direct glucuronide conjugation (T1) was observed in vitro in the HµREL system, and appears to represent a predominant metabolic pathway with continuous accumulation over 7 days (Fig. 1A). However, this conjugative metabolite was not reported previously in vivo (Tocco et al., 1980), which may be the result of advancement of analytical techniques or this metabolite being unstable or excreted into bile or feces and thus not identified in early metabolite identification work. All observed metabolites, accurate mass information, and key fragment ions are summarized in Supplemental Table 1.
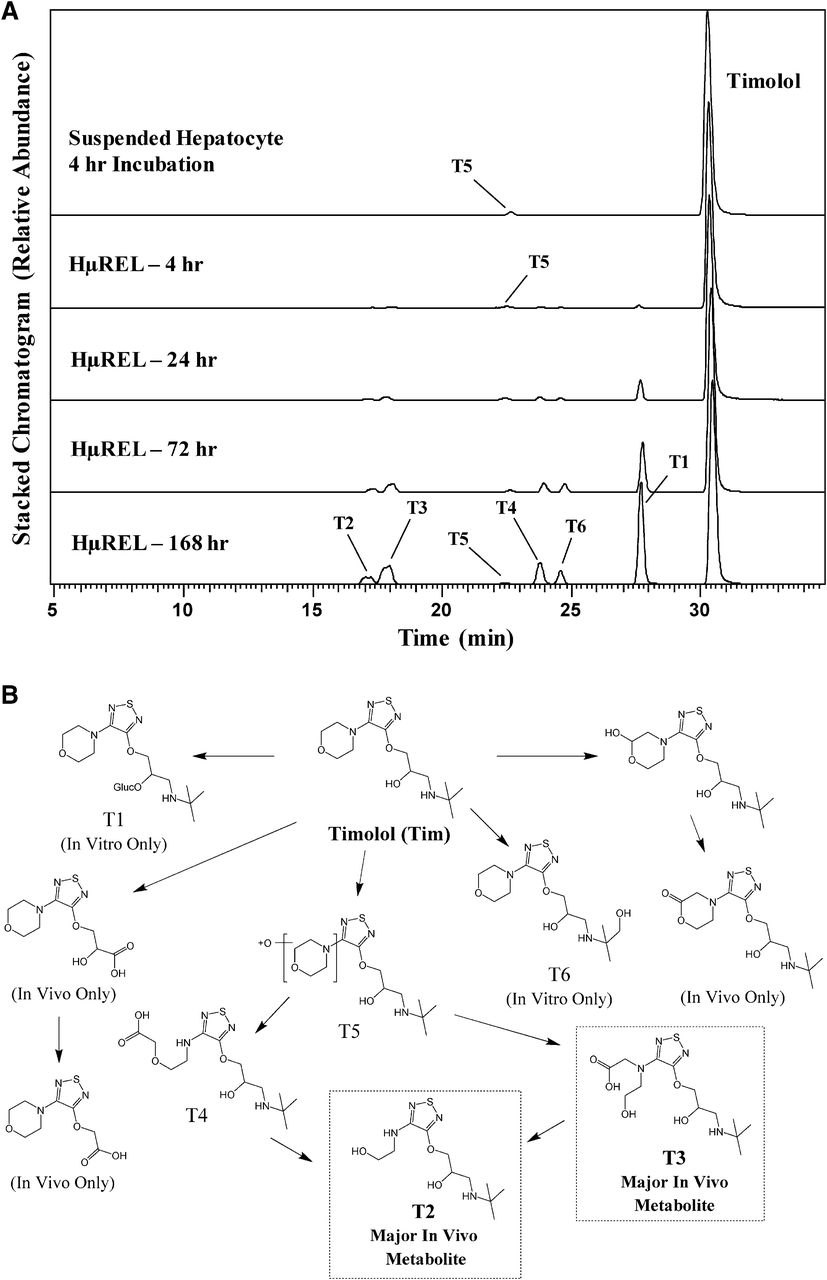
(A) Extracted ion chromatograms showing the metabolite profile of timolol following incubations in human suspended hepatocytes (4 hours) and the HµREL model (up to 7 days). (B) Proposed metabolic pathways of timolol. Metabolites highlighted in boxes are consistent with previously published human metabolite data. In vivo data were obtained from Tocco et al. (1980).
Meloxicam.
Profiling of drug metabolites following a 4-hour incubation of meloxicam with a conventional human hepatocyte suspension revealed minimal metabolic turnover with only parent drug and one minor metabolite (M5) being identified (Fig. 2A). The M5 metabolite was formed from hydroxylation of the methyl substituent of the methylthiazole moiety. Incubation of meloxicam utilizing the HμREL model generated a similar metabolic profile to the suspension hepatocytes following 4-hour incubation. More metabolites appeared at 24 hours and up to 7-day incubation with oxidation of the methylthiazole moiety representing the three most abundant metabolites (M4, M5, and M6). Two of these metabolites exhibited oxidation of the methyl group in the form of hydroxylation (M5) and the corresponding carboxylic acid (M4). Previously, M4 was found to be a major metabolite in human, representing approximately 60% of the dose (Schmid et al., 1995). Thus, the HµREL model more accurately predicted the in vivo metabolism of meloxicam than incubations using suspended hepatocytes (Fig. 2B). The metabolic profile for the HµREL model at 4 hours was comparable to that observed for the conventional suspension hepatocytes; suggesting that the two hepatocyte systems had similar biologic function in drug metabolism and the higher turnover observed from 24 hours to 7 days was mostly due to the longer-term viability of the hepatocytes in the HµREL system. Since M4 was the secondary metabolite of M5, its formation required a longer incubation time. Consequently, it was not observed at 4-hour incubation in both systems and was first observed at 24 hours in the HµREL model. Additional minor metabolites were observed resulting from hydroxylation (M3), glucuronide conjugation (M1), and oxidative cleavage (M2). All observed metabolites, accurate mass information, and key fragment ions are summarized in Supplemental Table 1.

(A) Extracted ion chromatograms showing the metabolite profile of meloxicam following incubations in human suspended hepatocytes (4 hours) and the HµREL model (up to 7 days). (B) Proposed metabolic pathways of meloxicam. Metabolites highlighted in boxes are consistent with previously published human metabolite data. In vivo data were obtained from Schmid et al. (1995).
Linezolid.
Incubation of linezolid using the conventional hepatocyte approach yielded only two minor metabolites, L1 and L2, which resulted from oxidative ring opening of the morpholine moiety. The metabolic profile was similar to that generated in the HµREL model following incubation for 4 hours. When the incubation time was prolonged, both metabolites were consistently produced and accumulated in the HµREL model. At day 7, the two metabolites were predominant and detected at a much higher level than those observed following 4-hour incubation, with L1 being more abundant than L2 (Fig. 3A). Consistently, L1 and L2 were reported to be prominent in human following oral dose of linezolid (Slatter et al., 2001), representing 45% and 11%, respectively, of the dose recovered in excreta at steady state. Additional metabolites (L3, L4, and L5) via the opening of the morpholine ring were observed in the HµREL model but not in suspended hepatocytes, as shown in Fig. 3B. These metabolites were also observed in a human in vivo study (Slatter et al., 2001). Therefore, the HµREL model again more closely predicted the in vivo metabolism of linezolid relative to hepatocytes in suspension. Interestingly, the similar metabolic profile of linezolid at day 7 was observed in another long-lived hepatocyte model, micropatterned hepatocyte coculture (Wang et al., 2010). This comparable result seems to suggest that the reason for generating more robust metabolites in the coculture models than suspension hepatocytes is driven by the longer action on the drug by the enzymes in the system. All observed metabolites, accurate mass information, and key fragment ions are summarized in Supplemental Table 1.

(A) Extracted ion chromatograms showing the metabolite profile of linezolid following incubations in human suspended hepatocytes (4 hours) and the HµREL model (up to 7 days). (B) Proposed metabolic pathways of linezolid. Metabolites highlighted in boxes are consistent with previously published human metabolite data. In vivo data were obtained from Slatter et al. (2001).
XK469.
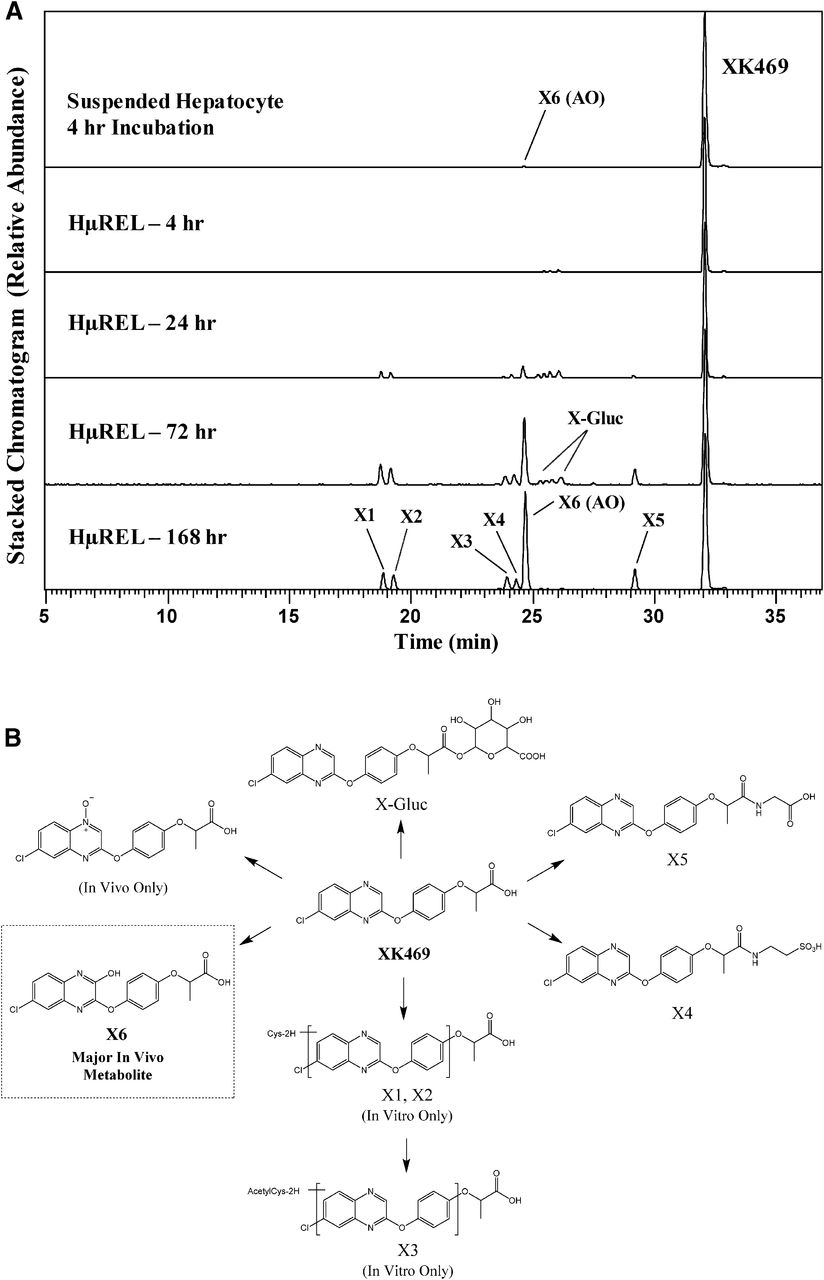
Incubation of XK469 in cryopreserved pooled human hepatocyte suspension for 4 hours produced only a single, low-abundant metabolite (X6), while the HμREL model produced robust metabolic turnover by 24 hours to a variety of metabolites covering both oxidative and conjugative pathways, with viable metabolism observed for up to 7 days (Fig. 4). The X6 metabolite was the major metabolite detected at days 3 and 7, which was produced as a result of mono-oxidation of the quinoxaline moiety. The oxidation on this position of the XK469 molecule is reported to be catalyzed by a nucleophilic mechanism via the molybdenum hydroxylase enzyme aldehyde oxidase (AO) (Anderson et al., 2005; Hutzler et al., 2012). This AO-derived metabolite was observed to be a major metabolite in human (Anderson et al., 2005), accounting for 54% of total urinary excretion. In addition, taurine (X4), glycine (X5), and glucuronide conjugates (X-Gluc) were produced following incubation of XK469 in the HμREL model, which were all observed previously in human, albeit at low levels. One metabolite reported in human, an N-oxide, was not observed following incubations using either the conventional hepatocyte suspension or HμREL model. Cysteine (X1 and X2) and acetylcysteine (X3) conjugates produced via the HμREL platform were not previously reported in human. Overall, the HµREL coculture system produced a metabolite profile that was much more complete and consistent with the human in vivo profile of XK469. All observed metabolites, accurate mass information, and key fragment ions are summarized in Supplemental Table 1.

(A) Extracted ion chromatograms showing the metabolite profile of XK469 following incubations in human suspended hepatocytes (4 hours) and the HµREL model (up to 7 days). (B) Proposed metabolic pathways of XK469. Metabolites highlighted in boxes are consistent with previously published human metabolite data. In vivo data were obtained from Anderson et al. (2005).
While metabolite identification studies were only conducted with the selected test molecules in human hepatocytes using the HµREL model, XK469 represents a good example of a drug that is slowly, but extensively, metabolized by multiple pathways, including AO (Anderson et al., 2005; Hutzler et al., 2012), which demonstrates profound species differences in expression and activity (Hutzler et al., 2013). If an uncharacterized novel drug candidate with similar metabolic properties to XK469 (i.e., extremely slowly metabolized) was to be incubated in a standard 4-hour human hepatocyte suspension, then an in vivo relevant metabolic pathway may be completely missed or underestimated. In addition, possible species differences in metabolism are less likely to be uncovered for slowly metabolized drugs in standard suspension incubations of limited duration (e.g., 4 hours). In the case of XK469, the AO pathway represents the predominant metabolic route, which is important to realize prior to regulatory in vivo toxicology studies and certainly prior to phase I human studies. SGX523 was an oncology drug candidate that was predominantly metabolized by AO to an insoluble metabolite that crystallized in the kidney and led to acute renal toxicity in human patients (Diamond et al., 2010). The AO pathway was completely missed in this example due to in vitro studies only being conducted in microsomal fractions (devoid of AO) as well as in toxicology studies being conducted in rat and dog, which are poor surrogates for human AO activity. However, the same scenario could be realized for a slowly metabolized AO substrate unless incubations are conducted for a long enough period to robustly generate metabolites for a clearer picture of the possible species-specific metabolic fate of a drug candidate.
Conclusions
In our studies evaluating four low-turnover drugs, the HµREL coculture model was able to generate a more robust metabolite profile over 7 days than that observed following incubation using conventional hepatocyte suspension. There were only one or two minor metabolites detected following suspension hepatocyte incubations with timolol, meloxicam, linezolid, and XK469 (Figs. 1–4). In comparison, multiple relevant human metabolites were detected in the corresponding HµREL incubations (Figs. 1–4), indicating that key metabolites are less likely to be missed during metabolite identification studies in a long-term coculture model such as HμREL. For example, the human predominant in vivo metabolites including T2, T3, M4, M5, L1, L2, and X6 (AO) were all observed at an appreciable level in the HµREL model (Figs. 1–4; Table 1). However, T2, T3, and M4 were not detected in the 4-hour suspension hepatocytes, likely because secondary and tertiary metabolites require longer exposure of the primary metabolites in the in vitro system. Collectively, data from these studies suggest that the HµREL model maintains metabolic activity over a diverse array of metabolic enzymes, and is a more ideal in vitro system than the conventional hepatocyte suspension in terms of in vivo metabolism prediction for low-turnover compounds or compounds that undergo multistep biotransformation.
Summary of metabolites observed following incubation in human hepatocyte suspension and the HµREL coculture model and comparison with reported in vivo metabolite data
While our studies only involved assessment of human metabolism, the HµREL model has multiple species of pooled hepatocytes to choose from to accommodate different study designs for species comparisons. Some conveniences of this model in comparison with other in vitro methods are worth consideration. Hepatocytes used in the coculture system are cultured by HμREL Corporation according to the client-preferred plate design for 7 days (acclimation period for peak enzyme activity) prior to being shipped to the laboratory. Upon receipt, the incubation of test articles can be initiated in 4 hours (following media change and acclimation period), and metabolite formation can be assessed at preferred times for up to 7 days. Thus, the system is more flexible and less labor intensive than other reported methodologies such as the relay method, which requires daily hepatocyte thawing and resuspension of supernatants from the previous day. Another advantage over other coculture models is that pooled hepatocytes can be plated in the HμREL model, whereas other models such as HepatoPac require plating of individual donors (Chan et al., 2013), which will lead to increased samples for analysis if multiple individual donors need to be evaluated. Nonetheless, just like any in vitro hepatocyte model, the metabolite data produced in the HμREL model is ultimately dependent on the enzyme activity in the hepatocyte pools selected. The next focus of studies in our laboratory will be to assess the suitability of the HμREL model for enzyme phenotyping, an additional major challenge for drugs that are slowly metabolized and require longer incubation time.
Authorship Contributions
Participated in research design: Burton, Hieronymus, Chamem, Heim, Anderson, Hutzler.
Conducted experiments: Hieronymus, Chamem, Heim.
Performed data analysis: Burton, Heim, Anderson.
Wrote or contributed to the writing of the manuscript: Burton, Hieronymus, Chamem, Heim, Anderson, Zhu, Hutzler.
Footnotes
- Received June 1, 2018.
- Accepted August 15, 2018.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AO
- aldehyde oxidase
- DDA
- data-dependent acquisition
- DIA
- data-independent acquisition
- HRMS
- high-resolution mass spectrometry
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}