


Abstract
CYP3A4 induction is not generally considered to be a concern for safety; however, serious therapeutic failures can occur with drugs whose exposure is lower as a result of more rapid metabolic clearance due to induction. Despite the potential therapeutic consequences of induction, little progress has been made in quantitative predictions of CYP3A4 induction-mediated drug-drug interactions (DDIs) from in vitro data. In the present study, predictive models have been developed to facilitate extrapolation of CYP3A4 induction measured in vitro to human clinical DDIs. The following parameters were incorporated into the DDI predictions: 1) EC50 and Emax of CYP3A4 induction in primary hepatocytes; 2) fractions unbound of the inducers in human plasma (fu, p) and hepatocytes (fu, hept); 3) relevant clinical in vivo concentrations of the inducers ([Ind]max, ss); and 4) fractions of the victim drugs cleared by CYP3A4 (fm, CYP3A4). The values for [Ind]max, ss and fm, CYP3A4 were obtained from clinical reports of CYP3A4 induction and inhibition, respectively. Exposure differences of the affected drugs in the presence and absence of the six individual inducers (bosentan, carbamazepine, dexamethasone, efavirenz, phenobarbital, and rifampicin) were predicted from the in vitro data and then correlated with those reported clinically (n = 103). The best correlation was observed (R2 = 0.624 and 0.578 from two hepatocyte donors) when fu, p and fu, hept were included in the predictions. Factors that could cause over- or underpredictions (potential outliers) of the DDIs were also analyzed. Collectively, these predictive models could add value to the assessment of risks associated with CYP3A4 induction-based DDIs by enabling their determination in the early stages of drug development.
Drug drug interactions (DDIs) are a major source of clinical problems leading either to severe adverse drug reactions (Honig et al., 1993; Gomez et al., 1995; Floren et al., 1997; Greenblatt et al., 1998; Backman et al., 2002, 2006) or a reduction in pharmacological effects (Back et al., 1979; Daniels et al., 1984; Heimark et al., 1987). The most common DDIs are associated with alterations in drug clearance, primarily due to inhibition of drug-metabolizing enzymes (DMEs), particularly cytochromes P450 (P450s). Predictions of in vivo DDIs based on in vitro drug metabolism data are increasingly being integrated into decision making about the development of potential drugs during the early stages of drug discovery. The success in these predictions is due largely to an increased understanding of DMEs at the molecular level, which has provided insight into the mechanisms of drug biotransformation and disposition. Substantial progress has been made in extrapolating in vitro inhibition data for the prediction of clinical DDIs, but fewer advances in prediction of induction-based DDIs have been realized.
Induction of CYP3A4 is especially important because the enzyme is involved in the metabolism of approximately 50% of marketed drugs (Guengerich, 1999). Unlike P450 inhibition, CYP3A4 induction is not as frequent a problem and is not normally thought to be a safety concern. Induction can lead to inadequate efficacy of coadministered drugs, and it is therefore an undesirable property (Dickinson et al., 2001; Grub et al., 2001). Induction studies have generally been more difficult to conduct experimentally than inhibition studies because induction is an indirect and slow process of gene up-regulation and increased protein expression; however, the advent of newer technologies such as nuclear receptor-reporter and mRNA analyses has partially overcome this problem. Although the concept of induction of DMEs has been known for several decades (Conney, 1967), an understanding of the mechanisms of P450 induction has only been developed slowly. The major mechanism of CYP3A4 induction has been determined to occur via activation of a human orphan nuclear receptor known as the pregnane X receptor (PXR) (Kliewer et al., 1998). Inducer binds to PXR in the cytosol and together they translocate to the nucleus where a heterodimer with retinoid X-receptor is formed. The receptor complex binds to the DNA-responsive element (DNARE) upstream of the CYP3A4 gene and activates its promoter, leading to transcription of the CYP3A4 gene. PXR also induces the transcription of a number of other drug-metabolizing enzymes including less potent induction of CYP2B and CYP2C9 (Kliewer et al., 1998). The extent of induction depends on the concentration of the inducer and on the duration of exposure.
Induction has been well characterized with a number of drugs, such as rifampicin (Backman et al., 1996), phenobarbital (Rutledge et al., 1988), troglitazone (Prueksaritanont et al., 2001), and bosentan (van Giersbergen et al., 2002), examples that are mainly confined to induction of CYP3A4. CYP3A4 is induced by a wide range of structurally diverse compounds that reflect a broad range of binding specificity for PXR. Inducers of CYP3A4 also induce CYP3A in other species but with major differences in the degree of response (Jones et al., 2000; Moore and Kliewer, 2000). For example, the prototypical CYP3A4 inducer rifampicin is a potent activator of human and rabbit PXR but has little activity on the rat and mouse receptor (Jones et al., 2000; Vignati et al., 2004). These species differences are known to be substantial, both in the spectrum of the enzymes induced and the extent of induction, and make nonhuman in vivo models inappropriate for induction studies intended to understand the risk of human induction. Because of these limitations, there is an increasing demand for the use of human in vitro systems (Masimirembwa et al., 2001). A commonly used technique is based on immortalized cell lines with engineered receptor and reporter systems (Goodwin et al., 1999). Another emerging technique is the use of immortalized human hepatocytes (Mills et al., 2004; Ripp et al., 2006). Despite these advances, primary human hepatocyte cultures have been and remain the gold standard in vitro system for investigating potential P450 induction (LeCluyse et al., 2000; Hewitt et al., 2007). Induction can be assessed with these cultures by measurements of both mRNA level and P450 functional enzymatic activity using probe substrates. The hepatocyte-based assays are, however, relatively low throughput and impractical for screening of large numbers of compounds because of the complexity of the assays and the need for a continuous supply of hepatocytes. Comparisons of induction measured in cell-based reporter assays and human hepatocytes have shown some degree of correlation (Luo et al., 2002).
The importance of CYP3A4 induction has prompted the search for alternative approaches to predicting DDIs using in vitro data obtained during the drug discovery stage. Unfortunately, little progress has been made in this regard. Development of predictive models of DDIs due to P450 inhibition have been based on fundamental principles and assumptions of in vitro in vivo extrapolations (IVIVE) and have been essential for predicting the magnitude of clinical DDIs. The success of these models demonstrates that in vitro data can be integrated into predictive models for in vivo DDI predictions and that useful predictions can be performed relatively early in the drug discovery process. These predictions aid in 1) selecting compounds for further development, 2) developing structure-activity relationships to avoid the potential for DDIs, and 3) planning of clinical DDI studies for compounds that are advanced into further drug development. In the present study, predictive models were developed and used for IVIVE of induction-based DDIs. Correlations of the predicted DDIs with observed in vivo DDIs were also analyzed.
Materials and Methods
Materials. The test articles were obtained from the following sources: dimethyl sulfoxide (DMSO), rifampin (RIF), phenobarbital (PB), dexamethasone (DEX), testosterone, 6β-hydroxytestosterone, Dulbecco's modified Eagle's medium (plating medium), and serum-free Williams' Medium E (culture medium) from Sigma-Aldrich (St. Louis, MO); bosentan (BST) and efavirenz (EFA) from BIOMOL International LP (Plymouth Meeting, PA); carbamazepine (CMZ) from MP Biomedicals (Solon, OH); fetal bovine serum from Invitrogen (Carlsbad, CA); sandwich medium from BD Biosciences (Bedford, MA); and Krebs-Henseleit buffer (KHB) from Amgen (Thousand Oaks, CA). Freshly isolated human hepatocytes were purchased from CellzDirect (Pittsboro, NC).
Human Hepatocyte Culture and Experimental Procedure. Fresh human hepatocytes from two donors were purchased from CellzDirect: donor 1, a 36-year-old white female (height 5 ft 3 in., body weight 160 lb, lot 0624) with no record of medications or of substance or alcohol abuse; and donor 2, a 41-year-old white female (height 5 ft 7 in., body weight 152 lb, lot 0697) with a history of smoking but no alcohol abuse. On day 1, fresh hepatocytes were received and suspended in plating medium (0.75 × 106 cells/ml). Hepatocytes were counted and plated in collagen-coated 24-well plates with a density of 0.4 × 106 cells/well (BD Biosciences). The hepatocytes were placed in a 37°C incubator (Steri-Cult CO2 Incubator, model 3310; Thermo Fisher Scientific, Waltham, MA) under an atmosphere of 95% air/5% CO2 and 90% relative humidity and allowed a 3 to 5-h attachment period. After the attachment period, the plating medium and unattached cells were removed by aspiration, sandwich medium was applied (0.5 ml/well), and the cells were incubated overnight. On day 2, the sandwich medium was aspirated and culture medium (0.5 ml/well) was applied for an overnight acclimation period. On days 3 and 4, culture medium containing either DMSO (0.1%), BST (0.5-50 μM), CMZ (1.0-100 μM), DEX (1.0-100 μM), EFA (0.5-20 μM), PB (15.6-1000 μM), or RIF (0.05-5 μM) was applied on each day (0.5 ml/well). The test articles were prepared in DMSO stock solutions, resulting in final incubation concentrations of 0.1% DMSO. Compound treatment was maintained for a total of 48 h. On day 5, hepatocytes were gently washed three times with KHB (0.5 ml/well, 37°C) and allowed to acclimate for an additional 10 min. P450 enzyme activities were subsequently determined by the addition of the marker substrate testosterone (200 μM; CYP3A4) dissolved in KHB (0.5 ml/well, 37°C). After a 15-min incubation, the medium was removed and stored at -80°C until analyzed. Hepatocytes used for mRNA analysis were washed once with phosphate buffered saline (0.5 ml/well, 25°C) containing calcium and magnesium and aspirated. Lysis mixture (0.5 ml) (Panomics, Inc., Fremont, CA) was added to each well, and the mixture was then stored at -80°C until assayed.
mRNA Analysis. CYP3A4 mRNA content was determined with branched DNA (b-DNA) signal amplification technology using the Panomics Discover XL Kit (Panomics, Inc.) with assays performed according to the manufacturer's instructions. b-DNA probe sets containing capture extender, label extender, and blocking probes for human CYP3A4 and GAPDH were also purchased from Panomics, Inc. Plate washing steps were performed on an Elx405 automated microplate washer (BioTek Instruments, Inc., Winooski, VT), and luminescence was analyzed on a Luminoskan Ascent microplate luminometer (Thermo Labsystems, Helsinki, Finland). P450 mRNA levels were normalized to the mRNA levels of the housekeeping gene GAPDH.
Enzyme Activity. After the induction treatment period, the metabolism rate of the CYP3A4 marker substrate, testosterone, by the cell cultures was determined. Analysis and quantification of 6β-hydroxytestosterone, the major testosterone metabolite in hepatocyte cultures, was performed by liquid chromatography-tandem mass spectrometry on a system comprising a reverse-phase high-performance liquid chromatograph (Shimadzu, Kyoto, Japan) and a triple quadrupole mass spectrometer (API 5000; Applied Biosystems, Foster City, CA) using Turbo IonSpray (Applied Biosystems) via multiple reaction monitoring. Samples (25 μl) were loaded on a C18 column (Onyx Monolithic C18, 100 × 3.0 mm, P/No. CHO-8158; Phenomenex, Torrance, CA), and analytes were eluted with a linear gradient of mobile phase A (H2O with 0.1% acetic acid and 5% methanol) to B (H2O with 0.1% acetic acid and 95% methanol) in 4.6 min. The flow rate was 1 ml/min. The metabolite was quantified by comparison of peak area ratios of metabolite to internal standard (prazosin) to a standard curve prepared using authentic 6β-hydroxytestosterone.
Fractions Unbound in Plasma and Hepatocytes. Frozen human plasma was thawed, aliquots (2 ml) were preheated to 37°C, and then the individual inducers were incubated at 1 and 10 μM in polypropylene centrifuge tubes (Corning Inc., Corning, NY) for 15 min. After this incubation period, 800-μl aliquots (n = 2) were transferred into polyallomar tubes (Beckman Coulter, Fullerton, CA), and then the tubes were placed in a MLA-130 rotor and centrifuged at 16,128g for 3 h at 37°C in an Optima Max Ultracentrifuge (Beckman Instruments, Palo Alto, CA). Similarly, the inducers at varying concentrations (5 μM BST, 20 μM CMZ, 100 μM DEX, 10 μM EFA, 250 μM PB, and 1 μM RIF) were incubated with human hepatocytes at 37°C for 5 min. After incubation, the tubes were centrifuged at 500g for 5 min. Aliquots (0.1 ml; n = 3) of the supernatants (the unbound fractions) were then removed from each tube. To precipitate proteins and prepare the samples for analysis, 50 μl of aliquoted supernatant from the plasma or hepatocyte samples was mixed with 200 μl of acetonitrile containing 0.125 μg/ml internal standard (proprietary Amgen compound). The samples were thoroughly mixed by vortexing and centrifuged for 10 min at 405g, and then 200 μl of the resultant supernatant was transferred into 96-well plate. Solvent was removed under a stream of N2 gas, and the samples were reconstituted with 100 μl of 50% MeOH/H2O for liquid chromatography-tandem mass spectrometry analysis. Standard curves and quality controls were prepared in the same manner. Concentrations of analytes were determined using a standard curve prepared in the same matrix. The unbound fraction in plasma (fu, p) or hepatocytes (fu, hept) was determined as a ratio of the concentration measured relative to total concentration. Recovery of drug was also determined by measuring the initial concentration of drug in the plasma or hepatocytes and comparing it to the nominal concentrations. All measurements were conducted in triplicate.
Data Analysis. In vitro induction assays were performed in triplicate. Concentration-response data sets of each inducer for mRNA and enzyme activity were plotted and fitted to the Hill equation (eq. 1) using Sigmaplot 10.0 (Systat Software Inc., Chicago, IL),
where Emax is the maximum response (net maximum fold increase), EC50 is the inducer concentration at 50% Emax, [Ind] is the inducer concentration, and n is the sigmoidity of the fitted curve.
CYP3A4 Induction Modeling. Induction of CYP3A4 enzyme by an inducer in hepatocytes is described as a receptor-mediated (e.g., PXR) process that triggers transcriptional activation of the gene coding for the protein. The fraction of occupancy of the DNA promoter elements by inducer complex (inducer-receptor) is expressed by eq. 2 and related to the concentration of the inducer:
where FO is fractional occupancy, [Ind] is the inducer concentration, and [Ind · r · DNARE] is the concentration of inducer, receptor, and DNARE complex. [DNARE] is the response element concentration, n is the Hill coefficient, and EC50 is the concentration of inducer at 50% maximal effect of induction.
Hepatic CYP3A4 content in the absence of inducer is governed by the rate of de novo enzyme synthesis (K0) and the rate of enzyme degradation (Kdeg) as shown in eq. 3. At steady state, the rate of de novo biosynthesis of the enzyme equals the degradation rate (K0 = Kdeg · [E]ss). It follows that the enzyme content ([E]ss) and intrinsic clearance (CLint) of a substrate of the enzyme are described by eqs. 4 and 5, respectively,
where K0 and Kdeg are the rates of enzyme synthesis and degradation, respectively; [E]ss is the enzyme content at steady state; kcat is the rate constant for metabolism of a substrate by the enzyme, and Km is the substrate-enzyme dissociation constant.
When an inducer is present, the enzyme level is raised to a new steady-state level as described in eq. 6. At the new steady state the enzyme level ([E]′ss) is defined by eq. 7 and intrinsic clearance (CL′int) for a substrate is determined by eq. 8:


where Kind is the rate constant for enzyme induction, Kmax is the maximum velocity of enzyme synthesis, and [E]′ss is the enzyme content at steady state in the presence of an inducer. The ratio of CLint in the absence and presence of an inducer is expressed by eq. 9, where Emax is the maximum induction of enzyme (net fold increase of enzyme):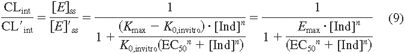
Prediction of DDIs from in Vitro CYP3A4 Induction. Based on pharmacokinetic principles, the AUC ratio of a substrate after intravenous administration in the presence and absence of an inducer is expressed by eq. 10. With oral administration the AUC of a substrate in the presence of an inducer is mainly affected by changes in hepatic bioavailability (F′h) and clearance (CL′h), assuming that gastrointestinal absorption and metabolic extraction (Eg) are not altered significantly [Fab = 1 (fraction of drug absorbed in gut) and Fg = 1 (gut bioavailability)] (eq. 11). Therefore, the AUC ratio of the dose is expressed in eq. 12: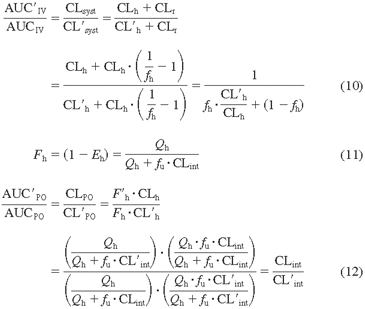
To predict the potential for drug interactions caused by CYP3A4 induction, a relationship of hepatic clearance to CYP3A4 intrinsic clearance (CLint, CYP3A4) is derived in eq. 13. If a compound is highly cleared by the liver (Qh ≪ fu · CLint or fu · CL′int) and CLh (or CL′h) is limited by the hepatic flow rate, then CLh is equal to CL′h (CLh/CL′h = 1). For a low-clearance drug (fu · CLint or fu · CL′int ≪ Qh), however, the ratio of CLh to CL′h can be expressed by eq. 13. The change in AUC ratio is then predicted by eq. 14 (eqs. 10 and 13 combined) for intravenous administration and by eq. 15 (eqs. 12 and 13 combined) for oral administration:

where [Ind] is the inducer plasma concentration achieved at steady state, usually Cmax, ss.
In many cases, Emax and EC50 are not readily obtained from concentration-response curves measured by in vitro induction assays because of limitations imposed by drug solubility, cell permeability, or toxicity. In these instances the slope of the induction response curve (equivalent to Emax/EC50) at a more experimentally feasible low concentration range of the inducer can be used for the prediction (eq. 16). This equation is, however, only applicable if in vivo concentrations of an inducer are low ([Ind] ≪ EC50). Eq. 17 shows the predictive model with inclusion of fractions unbound in plasma (fu, p) and hepatocytes (fu, hept), based on the hypothesis that only unbound drug (inducer) can access the intracellular nuclear receptor responsible for induction.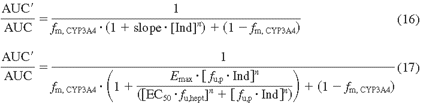
Contribution of CYP3A4 to Drug Clearance in Vivo (fm, CYP3A4). Fractions of the drugs cleared by CYP3A4 (fm, CYP3A4) were collected from clinical DDI studies reported in the literature. The extent of decreased clearance (or increased AUC) of a substrate drug in the presence of a selective CYP3A4 inhibitor (fi, CYP3A4) is used as an estimate of fm, CYP3A4 and calculated by eq. 18. In theory, an accurate value of fm, CYP3A4 should be given by the maximum decrease in clearance of a victim drug that is administered i.v. from clinical DDI studies (complete inhibition of the target enzyme by an inhibitor). However, most DDI studies reported in literature were performed by the oral administration of victim drugs, in which the first-pass metabolism of the drugs in the gut (Fg) could be affected (eq. 19) (Obach et al., 2006). Unfortunately, the estimated ratios of Fg′/Fg (Fg′ and Fg represent gut bioavailability in the presence and absence of inhibitor) in literature reports were not readily available for most drugs. Thus, the estimations of fm, CYP3A4 for the victim drugs of oral administration from clinical DDI data are based on assumptions that fg, CYP3A4 (eq. 19) is not changed in the presence of CYP3A4 inhibitor and that the inhibition of the CYP3A4 pathway is complete. In addition, when the fm, CYP3A4 values were calculated in the present study for the victim drugs given intravenously it is also assumed that the inhibition of intrinsic clearance of the CYP3A4 pathway by the inhibitors is complete (perhaps close enough for approximation), even for the high extraction drugs.
Correlation Analysis. The correlation analyses between predicted and clinical DDIs were performed by linear regression with analysis of variance. R2 (correlation coefficient) and residual sum of squares (RSS) (a measure of the size of the residuals, which are the differences of the actual data points from regression modeled values) were determined. In addition, weighted residuals (the residual divided by the standard error of the estimate) were analyzed to assess the distribution (normal or outlying) of the residuals.
Results
Concentration-Response Profiles of in Vitro CYP3A4 Induction. Human hepatocytes are routinely used as an experimental system for the evaluation of P450 induction potential. Selection of the model compounds, BST, CMZ, DEX, EFA, PB, and RIF, was based on 1) known CYP3A4 induction in vitro and in vivo, 2) minimal potential for mechanism-based inhibition of CYP3A4, and 3) known clinically relevant concentrations. Hepatocytes were prepared from fresh human livers (two donors), cultured for 2 days, and then treated with the inducers. After 48-h incubations with the inducers at varying concentrations, mRNA was analyzed using b-DNA technology, and CYP3A4 activity was determined by turnover of the marker substrate testosterone. The measured levels of induction in mRNA and enzyme activity were normalized against solvent control and recorded as the net fold increase over basal levels measured in control incubations. Full concentration-response curves were generated to provide complete kinetic characterization of each inducer (Fig. 1).
EC50 and Emax values for mRNA and enzyme activity were generated by fitting the observed data to the Hill equation (Table 1). All of the inducers were found to be capable of inducing CYP3A4 mRNA and activity. Moreover, there was general agreement between the two donors and between mRNA and activity parameters. As is commonly observed, however, some differences in the induction kinetics were observed. These differences occurred between the two donors and between the inductions measured by mRNA or activity levels. Generally, the enzyme activity Emax values for donor 1 were approximately 2- to 3-fold higher than those for donor 2, with the exception of RIF, which induced activity to a similar extent for both donors. Between the two donors, enzyme activity EC50 values for all of the inducers were similar with the exception of EFA, which had an approximately 5-fold higher EC50 in hepatocytes from donor 1 than that from donor 2. In hepatocytes from donor 1, the EC50 and Emax values for both mRNA and enzyme activity agreed within a 2-fold range except for a 6.7-fold difference in EC50 for EFA and a 4.7-fold difference in Emax for RIF. In donor 2 there was greater variability in both EC50 and Emax, and mRNA Emax values were generally higher than those for activity.
EC50 and Emax values of the inducers for CYP3A4 mRNA and activity in hepatocytes generated from the concentration vs. response curves in Fig. 1
S.D. shown in parentheses.
The greater CYP3A4 activity measured as testosterone 6β-hydroxylation after exposure to the inducers indicated that greater amounts of enzyme were present as a result of the induction. The relative in vitro induction potential of the inducers was also evaluated by determining changes in their intrinsic clearance (CLint = Emax/EC50 for enzyme activity). The analysis resulted in a rank order of RIF (42.0 and 24.5) > BST (19.6 and 4.1) > CMZ (1.03 and 0.17), EFA (0.58 and 1.45), DEX (0.33 and 0.25), and PB (∼0.1 and 0.07) for donors 1 and 2, respectively. The experimentally determined Emax and EC50 were incorporated into the prediction models together with fm, CYP3A4, clinical relevant concentration ([Ind]max, ss), protein binding in plasma (fu, p), and membrane partitioning in hepatocytes (fu, hept), as described below. Unbound fractions of the inducers in hepatocytes were measured to correct the EC50 values for free concentration as required for eq. 17. fu, hept was measured at inducer concentrations similar to their corresponding EC50 values in hepatocytes. The fractions unbound were widely variable among the compounds (Table 2). Interestingly, binding of the inducers to plasma and hepatocytes was observed to have a similar trend. As reported previously (Austin et al., 2002; Lobell and Sivarajah, 2003), this probably reflects a relationship between binding in either plasma or membranes and logD (or logP).
Unbound fractions of the inducers in human plasma and hepatocytes
Data are mean values in triplicate (S.D.).
CYP3A4 Reaction Phentotyping (fm, CYP3A4). The degree to which CYP3A4 contributes to the clearance of a drug, that is, the fraction of the drug metabolized by this isozyme (fm, CYP3A4), can be obtained from in vivo data (e.g., human radiolabeled compound disposition studies) or estimated by drug interaction studies with P450 isoform-selective inhibitors (fi, CYP3A4, eq. 18). fm, CYP3A4 is a critical parameter for the DDI prediction because, as shown in eq. 17, the magnitude of the DDI is directly proportional to this parameter. The precise role of an individual P450 in drug clearance, however, is difficult to ascertain because of multiplicity and overlapping substrate specificities for various P450 isoforms. In the present study, fm, CYP3A4 values were determined from clinical DDIs associated with CYP3A4 inhibition. The extent to which systemic or oral clearance of a drug is altered in the presence of an isozyme-specific inhibitor reveals the fraction of that drug cleared by CYP3A4. A number of CYP3A4-selective inhibitors have been widely used for coadministration with drugs suspected to be CYP3A4 substrates. These include the competitive inhibitors ketoconazole, fluconazole, itraconazole, and voriconazole and the mechanism-based inhibitors mibefradil, ritonavir, diltiazem, clarithromycin, and saquinavir (Table 3). The fm, CYP3A4 values were calculated by eq. 18 for the drugs used in this study and are shown in Table 3. As seen in Table 3, the fm, CYP3A4 values from many drug interaction studies ranged markedly from 0.05 (ropivacaine) to 0.95 (buspirone). High fractions indicate that the drugs are cleared mainly by CYP3A4, and low fractions imply that the drugs are cleared not only by CYP3A4 but also possibly by other routes including conjugative enzyme- or transporter-mediated pathways or renal or hepatic excretion.
Fractions of the drugs cleared by CYP3A4 (fm, CYP3A4) estimated by fi, CYP3A4 from clinical DDIs associated with CYP3A4 inhibition (eq. 18)

Curves of CYP3A4 response (fold increase) to inducer concentration (micromolar). IC50 and Emax values (Table 1) were generated by fitting of the observed data in hepatocytes to the Hill equation (eq. 1). Donor 1: A, mRNA; B, enzyme activity. Donor 2: C, mRNA; D, enzyme activity.
In Vivo Inducer Concentrations. To achieve a steady state of P450 induction, a human inducing agent must typically be chronically administered. Accordingly, for induction-mediated DDI studies subjects usually receive multiple doses of the inducer over consecutive days. After a steady state of induction is reached, the maximum concentration of the inducer ([Ind]max, ss) can then be measured. Ideally, for purposes of correlating a clinical DDI with [Ind]max, ss, the inducer concentration should be measured directly in the DDI study. In many of the studies cited herein, however, these measurements were not performed. The [Ind]max, ss values were therefore obtained from separate studies (Table 4) where the dose regimens were similar or identical to those used in the clinical DDI studies (Table 5). In the DDI studies the inducers were generally administered orally for several consecutive days followed by administration of the substrate drugs.
In vivo concentrations of the inducers at different dose regimens.
Comparisons of the predicted DDIs from the in vitro CYP3A4 induction in hepatocytes of the two liver donors (PRED1 and PRED2) with the clinical observed DDIs reported in cited references
In Vivo Drug-Drug Interactions. The decreased AUC ratios (AUC′/AUC) of the affected drugs in response to the inducers (Table 5) were reported within the cited references and resulted in 103 data points of drug/inducer interactions. The drugs represented in Table 5 are chemically and pharmacologically diverse with a wide range in magnitude of DDIs caused by CYP3A4 induction. This was therefore considered to be a suitable data set for the purposes of comparing actual DDIs with those predicted by eq. 17.
In Vitro-in Vivo Correlations. Various in vitro data were used for a model-directed prediction of DDIs with eq. 17. These data included the in vitro induction data (Table 1), unbound fractions of drug in hepatocyte incubations (fu, hept) and in plasma (fu, p) shown in Table 2, the fm, CYP3A4 values obtained from clinical CYP3A4 inhibition studies (Table 3), and the in vivo inducer concentrations at doses relevant to the DDI studies (Table 4). The predicted DDI values from the in vitro enzyme activity induction data in hepatocytes of the two donors for which fu, p and fu, hept of the inducers were included in the prediction are listed in Table 5. The measured clinical DDIs are also shown in Table 5, and these data sets (n = 103) and corresponding correlation analyses are shown in Figs. 2A and 3A. In addition to the predicted DDIs shown in Table 5 in which fractions unbound were included, correlation analyses were also performed without inclusion of the unbound fractions in the DDI prediction. These additional comparisons were performed to improve the understanding of the impact of these binding parameters on the correlations (Figs. 2, B-D, and 3, B-D). The correlation analyses included the 95% confidence interval (95% probability that the predicted DDI value will occur). A unity line (or unit correlation) is included in each figure to illustrate any difference from the observed correlation.
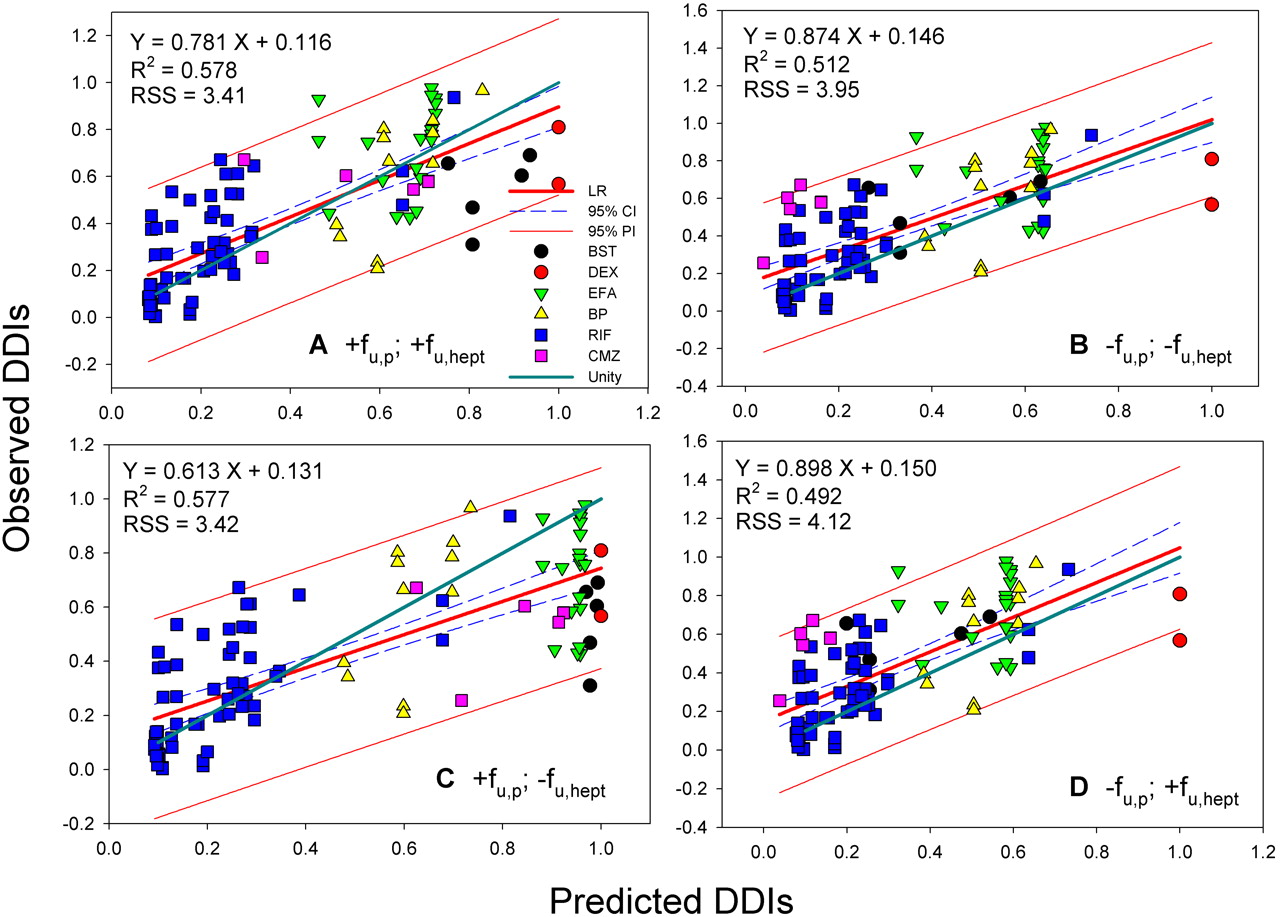
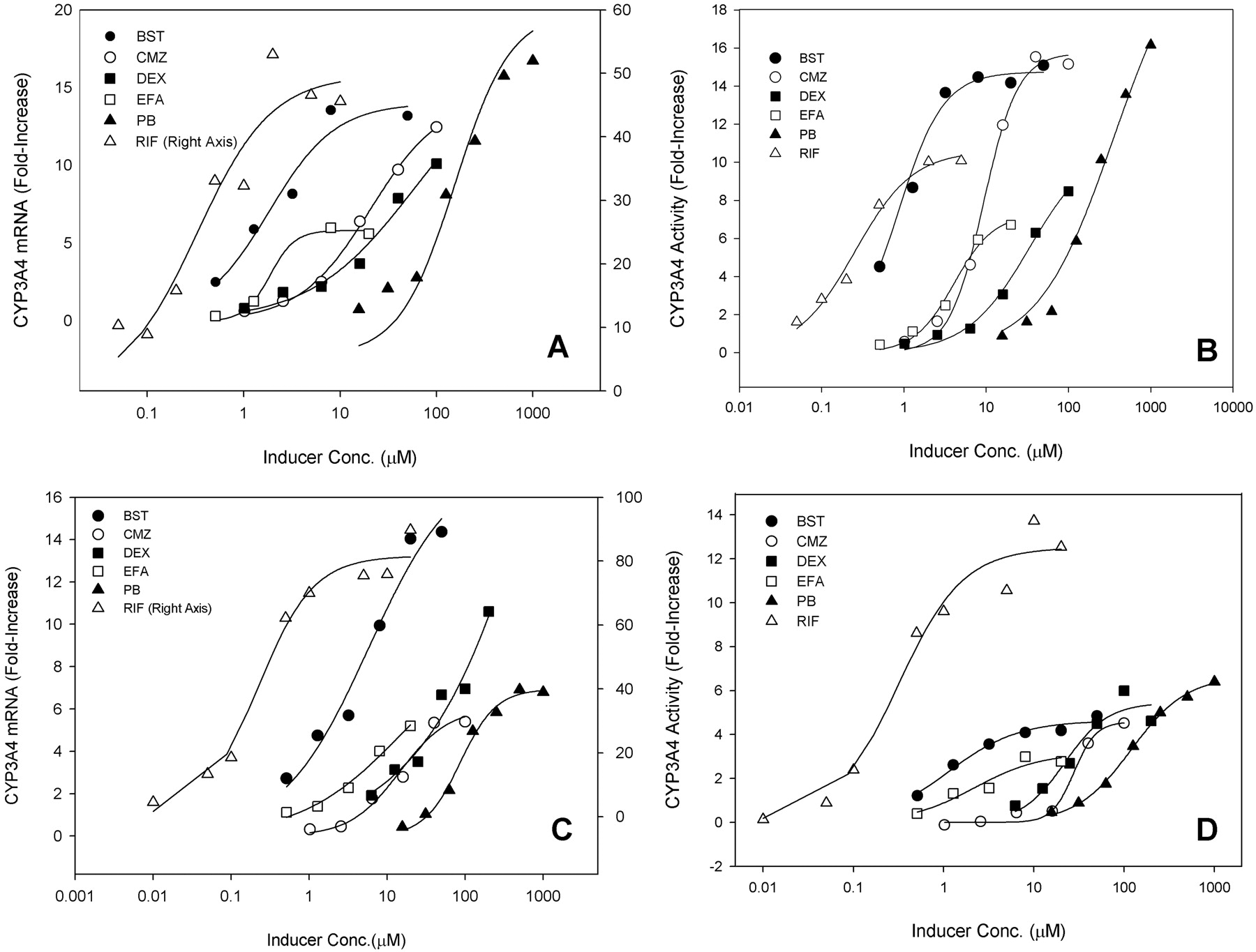
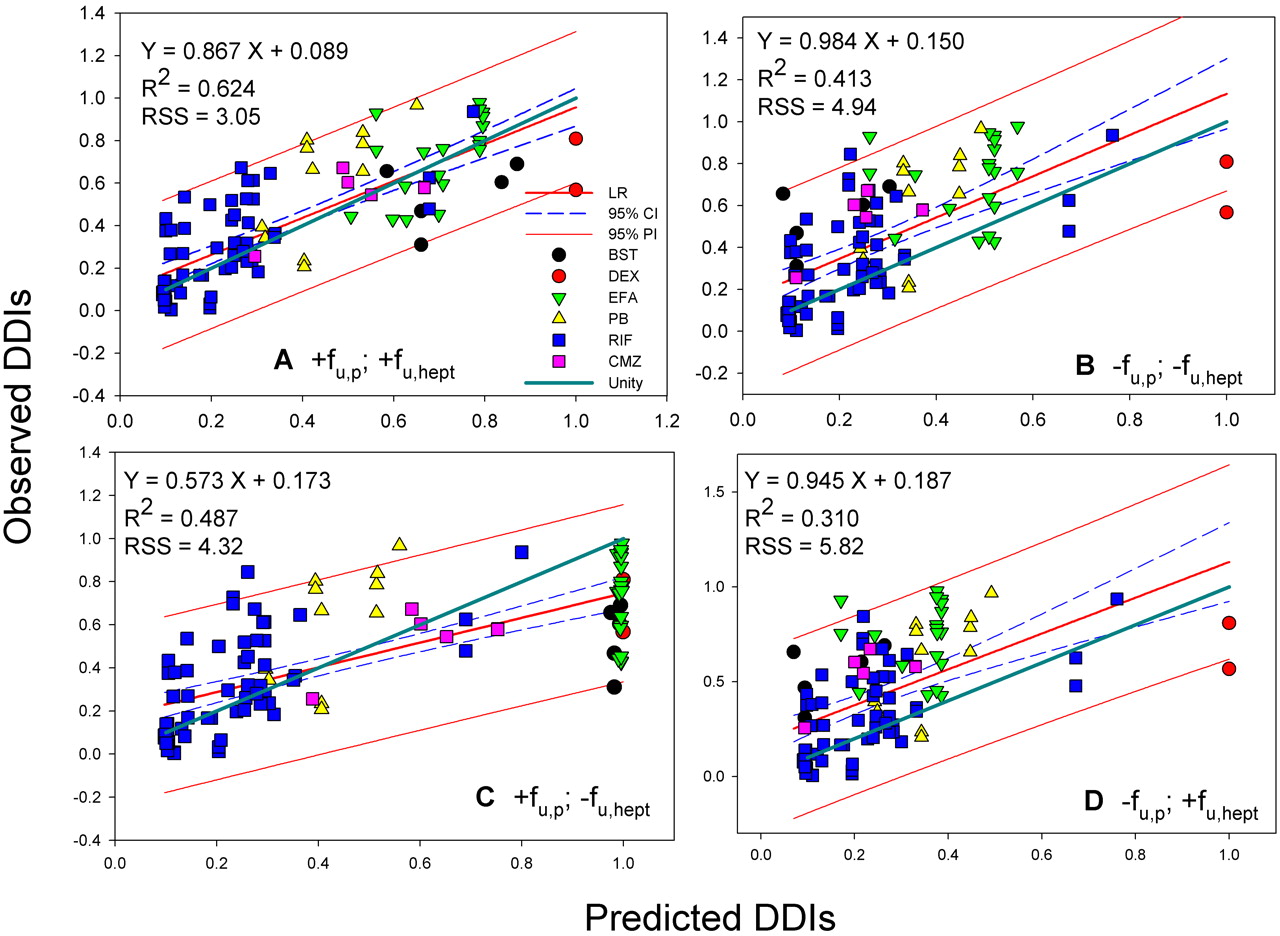
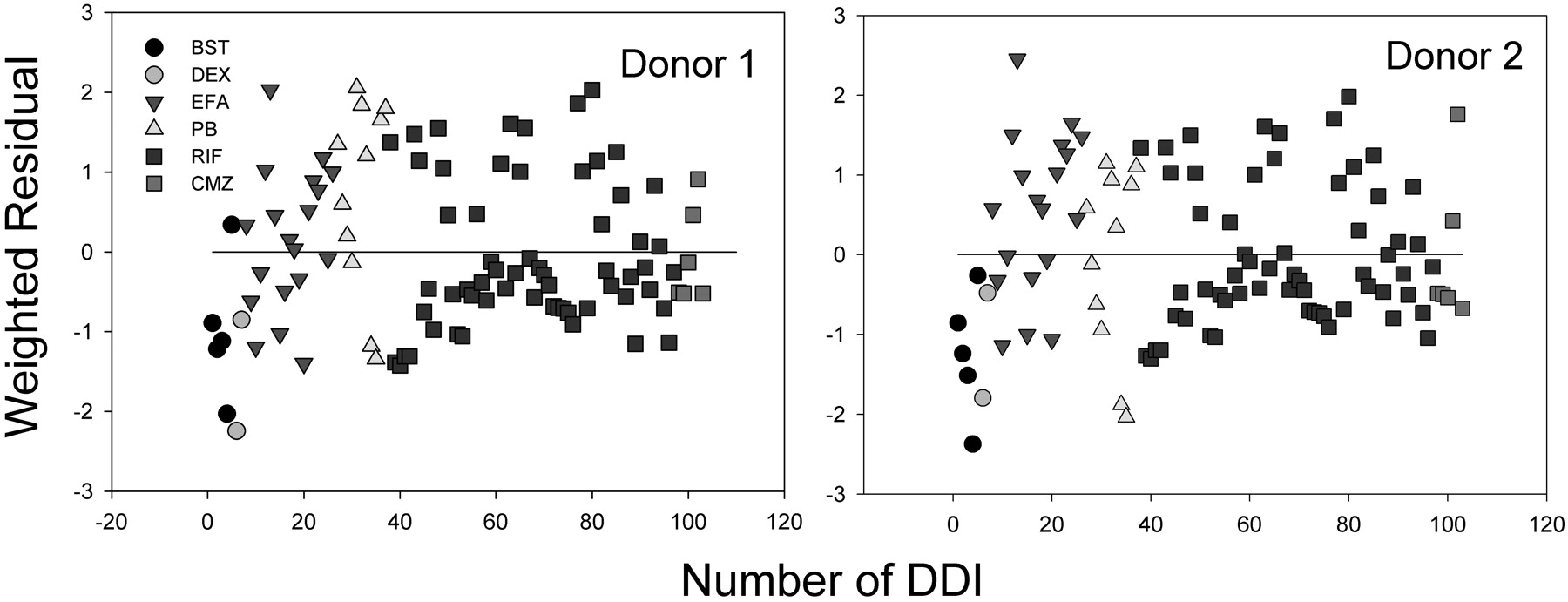
The best correlations were obtained when both fu, p and fu, hept were included in the analyses, leading to R2 = 0.624 for donor 1 (RSS = 3.05) and 0.578 for donor 2 (RSS = 3.41), respectively (Figs. 2A and 3A). The regression lines of correlation were very similar to the line of unity (unity correlation). When fu, p or fu, hept or both were excluded from the calculation, the correlations were found to be relatively poor (R2 = 0.310-0.577; RSS = 3.42-5.82) (Figs. 2, B-D, and 3, B-D). As might be expected, overpredictions of the DDIs were observed in the absence of the fu, p (Figs. 2, B and D, and 3, B and D). The degree of the overpredictions was dependent on the level of inducer binding to plasma. When fu, hept was ignored, the drug interactions appeared to be largely underpredicted, particularly for the inducers that were highly bound in hepatocytes such as EFA (Figs. 2C and 3C). Figure 4 shows the distributions of the weighted residuals of the correlations between the predicted and observed DDIs when both fu, p and fu, hept were included in the calculations. Most weighted residuals for the individual inducers were distributed around zero (unity regression) within ±2 units. The distribution ratios of positive to negative residual were 43:60 for donor 1 and 44:59 for donor 2, respectively. These show that the data were normally distributed around the unity.
When concentration-induction relationships are evaluated in hepatocytes, it is not uncommon to have difficulty determining EC50 and Emax values for P450 inducers that are poorly soluble or toxic. This difficulty arises because poor solubility and toxicity limit the ability to fully determine the concentration-response curves. This limitation can potentially be overcome by using the slope (Emax/EC50) of the induction-response curves obtained in the linear range at low concentrations. Using the slope, eq. 16 can then be used for the prediction. This approach is, however, only valid when in vivo concentrations of an inducer are much lower than its EC50, the conditions required for eq. 17 to be simplified to eq. 16. In the present study, EC50 and Emax values could be obtained for all of the inducers; however, it was observed that many of the in vivo concentrations of the inducers were higher than the EC50 values. For example, in vivo RIF concentrations were 10 to 18 μM (fu, p = 0.175), but the EC50 values for activity were 0.25 to 0.51 μM. When predictions by eq. 16 were attempted using the slope values for the inducers such as RIF, the DDIs were significantly overpredicted (data not shown). Predictions of drug interactions were also attempted from the in vitro EC50 and Emax values for mRNA induction shown in Table 1. The correlation analyses are shown in Fig. 5. The correlations were poor for both donors (R2 = 0.359 and 0.436), and for RIF in particular the DDIs were greatly overestimated. This overestimation was due predominantly to the much greater Emax observed for mRNA induction in both donors compared with enzyme activity.
Discussion
Marked induction of CYP3A4 by a drug is undesirable because of the large number of drugs that are dependent on CYP3A4 for their clearance. Induction by a drug can affect not only the clearance of a concomitantly administered medication but also its own clearance (autoinduction) by the induced enzyme (Tran-Johnson et al., 1987). Simple assays, such as reporter gene assays, that detect binding of inducers to receptors such as PXR have been used for measuring the capability of a drug to activate induction pathways; however, binding to PXR alone does not necessarily result in concomitant levels of enzyme induction (LeCluyse et al., 2000; Luo et al., 2002; Sinz et al., 2006). Fresh human hepatocytes maintained in primary culture are preferred for the direct assessment of the functional consequences of PXR activation, as measured by induction of mRNA and enzyme activity.
In the studies described here, differences in the kinetic characteristics of induction were observed between the two donors (Table 1). Such interindividual variability is not uncommon and imposes a degree of uncertainty in predicting the clinical consequences of induction. Differences in Emax values between mRNA induction and enzyme activity suggested that in some instances the parallel relationship between these two measures of induction can be altered by factors such as variations in rates of transcription, translation, and protein synthesis. Additional uncertainty in predictions also results from the metabolic instability of inducers in the cell cultures during the incubation time (48 h). Consumption of substrate could decrease the actual concentrations in the medium and thus differ from the nominal concentrations used for the calculation of kinetic parameters. In this regard, three inducers used here, BST (BST phenol metabolite), CMZ (CMZ-10,11-epoxide), and DEX (3-methoxymorphinan), have been reported to be CYP3A4 substrates (Gorski et al., 1994; Wang et al., 1997; Furukori et al., 1999; van Giersbergen et al., 2002; Kim et al., 2005).
In vivo fm, CYP3A4 is estimated by fi, CYP3A4 (eq. 18) as a change in the ratio of either AUC or clearance of a victim drug in the presence and absence of a coadministered CYP3A4 inhibitor (Brown et al., 2005; Venkatakrishnan and Obach, 2005; Galetin et al., 2006). The ratio ideally represents the proportion of the total clearance of a drug mediated by CYP3A4 at a relevant in vivo concentration of the inhibitor (Table 3). Under specific conditions fm, CYP3A4 will be equal to fi, CYP3A4; however, if those conditions are not met, fm, CYP3A4 could be either over- or underestimated. When a victim drug is administered orally and the AUC changes after inhibition, fm, CYP3A4 can be equal to fi, CYP3A4 only if the bioavailability (F) of the victim drug did not change. Any increase in F will result in a decrease in apparent clearance (CL/F), and fm, CYP3A4 will be overestimated compared with what would be derived from a true clearance value obtained if the victim drug was given intravenously. Many DDI studies are performed with oral dosing regimens, and, therefore, DDI predictions based on eq. 18 could be affected by potential errors of fm, CYP3A4 determined from fi, CYP3A4. This is particularly true for drugs with low bioavailability due to high first-pass extraction. In addition, some CYP3A4 inhibitors such as ketoconazole and indinavir also inhibit P-glycoprotein (P-gp). The inhibition of P-gp has been shown to increase oral absorption of substrates (Kim et al., 1998; Zhang et al., 1998; Achira et al., 1999) and would be another mechanism by which F could be affected.
Another requirement for fm, CYP3A4 to be equal to fi, CYP3A4 is complete inhibition of CYP3A4 in the inhibition studies in which fi, CYP3A4 is determined. Several of the inhibitors used to assess fm, CYP3A4 that are listed in Table 3 were recommended by the U.S. Food and Drug Administration draft guidance for use in DDI studies (http://www.fda.gov/cder/drug/drugInteractions/tableSubstrates.htm). These inhibitors were classified as either strong (ketoconazole, ritonavir, itraconazole, clarithromycin, and saquinavir) or moderate (fluconazole, grapefruit juice, and erythromycin) inhibitors. An assessment of whether CYP3A4 was indeed completely inhibited is dependent on the in vivo concentration (Cmax, ss) of the inhibitor that was achieved and the potency of the inhibitor (Ki, KI, and kinact). For example, the extent of inhibition achieved by ketoconazole (Cmax, ss = 8.9-13.1 μM, fu, p = 0.01, Ki = 0.015 μM, and ratio of unbound Cmax, ss/Ki = 5.9-8.7) (Varhe et al., 1994) and ritonavir (Cmax, ss = 7.8 μM, fu, p = 0.015, KI = 0.1 μM, and ratio of unbound Cmax, ss/Ki = 1.2) (Hsu et al., 1998) were likely to have been high. The inhibitors ritonavir, clarithromycin, saquinavir, and erythromycin have been shown to be potent and selective mechanism-based inhibitors of CYP3A4 (Greenblatt et al., 1999) and the extent of inhibition with these compounds was likely to have also been high. Mibefradil (KI = 2.3 μM and kinact = 0.4 min-1) has been shown to have an in vitro potency greater than clarithromycin (KI = 5.5 μM, kinact = 0.072 min-1) (Prueksaritanont et al., 1999), and clinical DDIs caused by mibefradil (1.2- to 7.8-fold) were comparable with those caused by clarithromycin (1.3- to 8.5-fold), depending on the extent to which the victim drugs were cleared by CYP3A4 (Jacobson, 2004). The final requirement for fm, CYP3A4 to be equal to fi, CYP3A4 is selective inhibition of CYP3A4 in the DDI clinical trial. The CYP3A4 selectivities of some inhibitors (e.g., ketoconazole and itraconazole) have been reported to be concentration- and substrate-dependent (Galetin et al., 2005). If other P450s responsible for the clearance of the victim drug are also inhibited, the fm, CYP3A4 can be overestimated.

Correlation analyses of predicted DDIs (Donor 1) with observed in vivo DDIs. A, with fu, p and fu, hept. B, without fu, p and fu, hept. C, with fu, p and without fu, hept. D, without fu, p and with fu, hept. LR, linear regression; R2, correlation coefficient; 95% CI, 95% confidence interval; 95% PI, 95% prediction interval; unity, unity correlation; RSS, residual sum of squares.
The dependence of DDI susceptibility on fm, CYP3A4 and [Ind]max, ss is illustrated in simulated RIF- and PB-mediated drug interactions, respectively (Fig. 6). Several laboratories have attempted to predict in vivo DDIs using a relative induction score (Ripp et al., 2006) or an induction ratio (Kato et al., 2005) measured in hepatocytes. In neither case, however, was fm, CYP3A4 incorporated into the models. Figure 6 clearly illustrates the importance of that parameter in any DDI prediction. The observed in vivo fm, CYP3A4 varied widely for the substrates examined, from 0.05 (ropivacaine) to 0.95 (buspirone). Low fm, CYP3A4 values suggest possible involvement of other clearance pathways of the drugs rather than CYP3A4, such as glucuronidation (ethinyl estradiol), CYP2C9 (losartan), and CYP2C9/19 and CYP2B6 (diazepam). Those drugs (e.g., alfentanil, simvastatin, and midazolam) with high fm, CYP3A4 values are cleared more extensively by CYP3A4, leading to a high degree of susceptibility to a DDI (Table 5). An example of this is the RIF-midazolam interactions, in which the midazolam AUC was reduced to approximately one-tenth the AUC in the noninduced state. Because of its potent enzyme induction, RIF caused large interactions with other drugs; however, the clinically observed decreases in exposure due to coadministration with RIF were greater than those predicted in many cases, particularly for drugs with relatively low fm, CYP3A4 values. This is because the CYP3A4-dependent portion of the total clearance of the drug increases upon induction. In addition, the clearance of drugs by CYP3A4 inducers such as RIF can be affected by increased expression of other enzymes and transporters regulated by PXR transactivation or other induction mechanisms such as CAR (Hewitt et al., 2007). For example, PB was only a moderate inducer of CYP3A4 in primary human hepatocytes and human PXR transactivation assays at its Cmax concentrations (Luo et al., 2002; Sinz et al., 2006), yet it has shown clinically significant DDIs (Table 5). This is probably because PB also induced CYP2B6 and CYP2C, contributing to the clearance of the affected drugs by its CAR-mediated induction and, to a lesser extent, PXR-mediated induction (Luo et al., 2002).
To predict the magnitude of in vivo DDIs, the most appropriate inducer concentrations to use in the calculation would be the concentration achieved in vivo at the target enzyme in the liver. Many drugs enter hepatocytes by passive diffusion, which allows projecting intracellular hepatic concentrations on the basis of systemic unbound concentrations ([Ind]max, ss · fu, p). This projection may not be appropriate if 1) the inducers are actively transported into or out of hepatocytes, 2) the inducers or affected drugs are inhibitors of transporters expressed in the liver that transport the inducers (e.g., EFA, verapamil, ritonavir, and cyclosporine), or 3) the inducers are extensively metabolized by CYP3A4 (e.g., DEX, BST, and CMZ) and the systemic concentration is merely a fraction of the liver exposure. Clearly there are multiple variables that can confound the assumptions in eq. 17, and, accordingly, under- or overprediction of the DDIs can be expected. For example, BST has been reported to be a substrate for hepatic OATP1B1 and OATP1B3 (Treiber et al., 2007). The underpredictions of the DDIs by BST with several CYP3A4 substrates, e.g., ethinyl estradiol, glyburide, and sildenafil were probably due to higher concentrations of the inducer in hepatocytes than in plasma. Similarly, RIF has been shown to be a substrate for and inhibitor of the uptake transporters OATP-C and OATP1B1 in the liver (Tirona et al., 2003). In addition, the liver inlet concentration of inducer after oral administration ([Ind]inlet, ss) is a function of the concentration in both the hepatic artery ([Ind]max, ss) and the portal vein after gut absorption (eq. 20):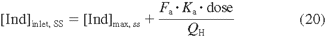
Correlation analyses of predicted DDIs (donor 2) with observed in vivo DDIs. A, with fu, p and fu, hept. B, without fu, p and fu, hept, C, with fu, p and without fu, hept. D, without fu, p and with fu, hept. LR, linear regression; R2, correlation coefficient; 95% CI, 95% confidence interval; 95% PI, 95% prediction interval; unity, unity correlation; RSS, residual sum of squares.

Weighted residual distributions (difference of the weighted residual from zero, the perfect linear regression) of the correlations (corrected by fu, p and fu, hept) between predicted and observed DDIs.

Correlations of the DDIs predicted from the in vitro mRNA induction data (including fu, p and fu, hept) in donors 1 (A) and 2 (B) with the clinically observed DDIs. LR, linear regression; R2, correlation coefficient; 95% CI, CYP3A4; 95% PI, 95% prediction interval; unity, unity correlation; RSS, residual sum of squares.

Simulated relationship of predicted DDIs as a function of in vivo free concentration of inducer ([Ind]max, ss) and fraction of drug cleared by CYP3A4 (fm, CYP3A4) using eq. 17. RIF (A and B): EC50 = 0.25 μM, Emax = 10.6 (fold increase), fu, p = 0.179 and fu, hept = 0.419; PB (C and D): EC50 = 250 μM, Emax = 22.6 (fold increase), fu, p = 0.7 and fu, hept = 1.0.
where Fa, Ka, and QH are the fraction of dose absorbed from the gut into the portal vein, absorption rate constant of inducer, and hepatic blood flow rate, respectively. For example, [Ind]max, ss (0.1 μM) of DEX is lower than the [Ind]inlet, ss (0.25 μM), calculated by eq. 20 with Ka = 2.68 h-1, Fa = 0.88, and dose = 1.5 mg (q.d., 4 days). This difference could in part explain the underpredictions of DEX interactions with cyclophosphamide and triazolam, respectively, in Table 5.
The gut may contribute a large portion of the first-pass extraction of CYP3A4 substrates, particularly for those substrates extensively cleared by that isozyme (Thummel et al., 1996). For example, the bioavailability (F) of cyclosporine was reduced by the coadministration of RIF (Ito et al., 1998) by increased metabolism in both gut and liver and also by induction of P-gp (Wacher et al., 1995). The latter highlights an additional complexity: RIF, PB, and DEX are also inducers of P-gp via the same nuclear receptor (PXR) that mediates the induction of CYP3A4 (Schuetz et al., 1996). Thus, the additive effects of increased CYP3A4-mediated metabolism and P-gp-mediated efflux by the intestinal epithelium may result in a substantially higher first-pass effect in the gut after oral administration. Without consideration of the gut first-pass effect, DDIs can be underpredicted. In addition, many drugs act as both inducers and inhibitors of CYP3A4 (ritonavir and EFA). When EFA was coadministered with ritonavir (KI = 0.04 μM), indinavir, or amprenavir (a potent mechanism-based inhibitor) in combination therapy (Luo et al., 2003), induction-mediated DDIs were overpredicted as a result of the potent CYP3A4 inhibition by the interacting drugs (Table 5).
A high degree of interindividual variability of DDIs has been commonly observed in the clinic. This variability could be due to genetic polymorphisms of P450s and transporters. For example, whereas CYP3A5 and CYP3A4 share common substrates and inducers, CYP3A5 is less inducible, differs in its tissue expression pattern, and is subject to genetic polymorphism (Kuehl et al., 2001). CYP3A4 can also vary in its abundance within a population. Combined, these factors result in the potential for a large degree of interindividual variability of DDIs after treatment with CYP3A inducers (Lamba et al., 2002). Similar genetic polymorphisms may also affect OATP-C drug disposition. OATP-C expression is potentiated by PXR activation; however, it has a number of naturally occurring variant forms that have markedly impaired function (Tirona et al., 2003).
In addition to the CYP3A4 induction-mediated DDIs described here, the predictive models may also be useful for the evaluation of potential DDIs from induction of other P450 isoforms such as CYP1A2, CYP2B6, and CYP2C9. The induction of these other P450s is regulated by different nuclear receptors (i.e., aryl hydrocarbon receptor and CAR); however, the fundamental principles of the potential of a drug to induce enzyme activity and mRNA in hepatocytes and the endpoint measurements of Emax and EC50 are identical to those used for CYP3A4.
The results of the analyses described here reveal some valuable insight into the correlation between in vitro induction and clinical DDIs. The draft U.S. Food and Drug Administration guidance for drug interaction studies (http://www.fda.gov/cber/guidelines.htm) recommends using induction of ≥40% of the positive control as one endpoint for prediction of clinically relevant enzyme induction. The absolute level of induction that a value of 40% represents is, of course, relative to the extent of induction caused by the positive control, which in turn is dependent on the potency of the positive control (Emax and EC50) and the concentration tested. The results described here show that 40% of Emax for the potent inducers RIF and PB corresponded to 4.2- and 9.1-fold increases in CYP3A4 activity in donor 1, respectively. The respective reductions of midazolam AUC predicted by eq. 17 for these levels of induction were 78 and 89%. This finding suggests that induction by a test drug that is 40% of the response for potent inducers that are tested at or near their Emax concentrations may result in an excessively large DDI.
The model has the potential to be used for classification of an inducer. For example, cutoffs that are proportionately similar to those used to rank P450 inhibitors might be applied. Using those criteria, induction could be based on the AUC in the induced state compared with the control and ranked as none (≥75%), weak (50-75%), moderate (20-50%), or strong (≤20%). A corresponding set of in vitro values of potency can be calculated by eq. 21 assuming fm, CYP3A4 → 1. Using this approach, induction of none, weak, moderate, and strong has a ratio of ≤0.33, 0.33 to 1.0, 1.0 to 4.0, and >4.0, respectively. Similarly, if Emax and EC50 for a drug cannot be obtained, the slope of the response-concentration curve in the linear range measured in hepatocytes can be also used, providing the projected [Ind] < EC50: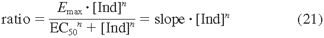
In conclusion, measurement of in vitro induction parameters for a drug candidate at an early stage of drug discovery can provide valuable information about the potential of a drug to cause clinical DDIs. If in vitro induction is observed and DDIs are predicted using the models described here, an informed decision about termination or advancement of the molecule can be made with a better understanding of the associated risk than has previously been available. Should the molecule be advanced, better planning and timing of clinical DDI studies may be possible. Confidence in the DDI prediction could allow for more rapid lead optimization by avoiding unnecessary optimization efforts or more rapid termination decisions. Ultimately, the decision of whether a drug candidate with a potential of induction should be advanced into development will depend on the magnitude of the predicted DDIs, the clinical indication, the dosing regimen, and concomitant medications that may be P450 inhibitors or predominantly cleared by the induced enzyme.
Acknowledgments
We thank Drs. Kenneth Korzekwa and Paul Pearson for valuable comments on the work.
Footnotes
-
doi:10.1124/dmd.108.020602.
-
ABBREVIATIONS: DDI, drug-drug interaction; DME, drug-metabolizing enzyme; P450, cytochrome P450; PXR, pregnane X receptor; DMSO, dimethyl sulfoxide; RIF, rifampin; PB, phenobarbital; DEX, dexamethasone; BST, bosentan; EFA, efavirenz; CMZ, carbamazepine; KHB, Krebs-Henseleit buffer; b-DNA, branched-DNA; GADPH, glyceraldehyde-3-phosphate dehydrogenase; AUC, area under the curve; RSS, residual sum of squares; P-gp, P-glycoprotein; CAR, constitutive androstane receptor; OATP, organic anion-transporting polypeptide; DNARE, DNA-responsive element; IVIVE, in vitro in vivo extrapolations.
- Received January 25, 2008.
- Accepted July 29, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}