


Abstract
As a direct-acting inhibitor of CYP2C19 in vitro, lansoprazole is more potent than omeprazole and other proton pump inhibitors (PPIs), but lansoprazole does not cause clinically significant inhibition of CYP2C19 whereas omeprazole does. To investigate this apparent paradox, we evaluated omeprazole, esomeprazole, R-omeprazole, lansoprazole, and pantoprazole for their ability to function as direct-acting and metabolism-dependent inhibitors (MDIs) of CYP2C19 in pooled human liver microsomes (HLM) as well as in cryopreserved hepatocytes and recombinant CYP2C19. In HLM, all PPIs were found to be direct-acting inhibitors of CYP2C19 with IC50 values varying from 1.2 μM [lansoprazole; maximum plasma concentration (Cmax) = 2.2 μM] to 93 μM (pantoprazole; Cmax = 6.5 μM). In addition, we identified omeprazole, esomeprazole, R-omeprazole, and omeprazole sulfone as MDIs of CYP2C19 (they caused IC50 shifts after a 30-min preincubation with NADPH-fortified HLM of 4.2-, 10-, 2.5-, and 3.2-fold, respectively), whereas lansoprazole and pantoprazole were not MDIs (IC50 shifts < 1.5-fold). The metabolism-dependent inhibition of CYP2C19 by omeprazole and esomeprazole was not reversed by ultracentrifugation, suggesting that the inhibition was irreversible (or quasi-irreversible), whereas ultracentrifugation largely reversed such effects of R-omeprazole. Under various conditions, omeprazole inactivated CYP2C19 with KI (inhibitor concentration that supports half the maximal rate of inactivation) values of 1.7 to 9.1 μM and kinact (maximal rate of enzyme inactivation) values of 0.041 to 0.046 min−1. This study identified omeprazole, and esomeprazole, but not R-omeprazole, lansoprazole, or pantoprazole, as irreversible (or quasi-irreversible) MDIs of CYP2C19. These results have important implications for the mechanism of the clinical interaction reported between omeprazole and clopidogrel, as well as other CYP2C19 substrates.
Introduction
Omeprazole and other proton pump inhibitors (PPIs, i.e., esomeprazole, lansoprazole, dexlansoprazole, rabeprazole, and pantoprazole) are well known for a relatively low incidence of adverse events and pharmacokinetic drug-drug interactions (DDIs). Nevertheless, PPIs are the perpetrators of interactions with cyclosporine (omeprazole and rabeprazole), diazepam (esomeprazole and omeprazole), and warfarin (esomeprazole, lansoprazole, omeprazole, and rabeprazole) (Wallace and Sharkey, 2011). In addition, all drugs that increase gastric pH can affect the bioavailability of drugs such as ampicillin esters, iron salts, and ketoconazole, among others (Wallace and Sharkey, 2011). Omeprazole, the first approved PPI, decreases the clearance of drugs such as diazepam, moclobemide, escitalopram, carbamazepine, saquinavir, sibutramine, proguanil, etravirine, disulfiram, phenytoin, voriconazole, and clopidogrel, and, by inducing CYP1A2, it increases the clearance of several antipsychotic drugs, such as imipramine, theophylline, and tacrine (Angiolillo et al., 2011; Wallace and Sharkey, 2011; MTDI database, http://www.druginteractioninfo.org).
The inhibitory DDIs listed above have been attributed, at least in part, to inhibition of CYP2C19 by omeprazole. However, clinically relevant DDIs with omeprazole are generally of low magnitude [≤120% (2.2-fold) increase in plasma AUC of CYP2C19 substrates; (MTDI database)]. By way of comparison, there is up to a 14.6-fold increase in the area under the curve (AUC) of omeprazole when CYP2C19 is completely absent as occurs in genetically determined CYP2C19 poor metabolizers (Furuta et al., 1999). Nearly 40 in vitro studies examining PPIs as P450 inhibitors have been published to date (MTDI database). Only omeprazole and lansoprazole (and their S-enantiomers) have been shown to inhibit CYP2C19, with IC50 or Ki values ≤1 μM. Of particular interest is that none of these in vitro studies examined the PPIs as metabolism-dependent inhibitors (MDIs; also known as time-dependent inhibitors) of cytochrome P450 (P450) enzymes. On the basis of the lowest reported Ki values for CYP2C19 inhibition in the MTDI database (i.e., 0.45 μM for lansoprazole and 1 μM for omeprazole) and the reported plasma Cmax for lansoprazole and omeprazole in CYP2C19-extensive metabolizers (2.2 and 3.9 μM, respectively; Li et al., 2004; Hassan-Alin et al., 2005), the [I]total/Ki values for lansoprazole and omeprazole would be 4.9 and 3.9, respectively [calculated as recommended in the Draft U.S. Food and Drug Administration (FDA) Guidance for Industry, 2006, http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm080499.htm], which means that both drugs would be expected to cause clinically relevant direct inhibition of CYP2C19, but that lansoprazole would have a higher likelihood of doing so than would omeprazole. However, lansoprazole has been reported to cause no interaction with the CYP2C19 substrates diazepam and phenytoin (MTDI database, Wallace and Sharkey, 2011). The lack of clinically relevant direct inhibition of CYP2C19 by lansoprazole is likely explained by its relatively short half-life (1.1 h; Li et al., 2004) and high plasma protein binding (∼97%; Prevacid prescribing information, 2010, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020406s074,021428s021lbl.pdf). However, omeprazole also has a short half-life (0.7 h; Hassan-Alin et al., 2005) and high plasma protein binding (∼95%; Prilosec prescribing information, 2011, http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/019810s092,022056s008lbl.pdf), and yet, as described above, omeprazole does cause clinically significant inhibition of CYP2C19. This apparent discrepancy could be explained if omeprazole, but not lansoprazole, were an irreversible inhibitor of CYP2C19.
In support of this possibility, we presented preliminary in vitro evidence for metabolism-dependent inhibition of CYP2C19 by omeprazole (Paris et al., 2008). More recently, a large clinical study in 282 healthy subjects (Angiolillo et al., 2011) demonstrated that omeprazole inhibited the CYP2C19-dependent activation of clopidogrel as evidenced by a 40 to 47% decrease in the formation of H4, the purported active antiplatelet metabolite of clopidogrel. Inhibition of clopidogrel activation was observed even when the two drugs were administered 12 h apart. In the same subjects, no such interaction was observed between pantoprazole and clopidogrel.
The interaction between clopidogrel and PPIs and the effect of the CYP2C19 poor metabolizer phenotype have prompted warnings from the FDA and European Medicines Energy (2010, http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm203888.htm; http://www.ema.europa.eu/docs/en_GB/document_library/Public_statement/2010/03/WC500076346.pdf), although the FDA specifically warns against coadministration of clopidogrel and omeprazole and further specifically suggests that pantoprazole may be a safer alternative (2010, http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm).
The identity of the enzyme(s) responsible for the multistep activation of clopidogrel is controversial (Savi et al., 1994; Clarke and Waskell, 2003; Abraham et al., 2010; Kazui et al., 2010; Zahno et al., 2010). Much of the pharmacogenomic data strongly implicate CYP2C19 as the most important enzyme for clopidogrel activation on the basis of the poor response to clopidogrel in carriers of reduced-function CYP2C19 alleles (e.g., the *2 and *3 alleles) and increased bleeding in carriers of the increased-function CYP2C19*17 allele (Sibbing et al., 2010). The DDI between PPIs and clopidogrel is of particular importance because PPIs are coprescribed in up to approximately two thirds of patients after discharge from the hospital because PPIs lessen the severity of the gastrointestinal hemorrhage associated with clopidogrel treatment (Ho et al., 2009).
In the present study, we examined the in vitro inhibitory potential of omeprazole, its individual enantiomers and selected metabolites, as well as lansoprazole and pantoprazole (Fig. 1) in pooled human liver microsomes (HLM), pooled cryopreserved hepatocytes, and recombinant CYP2C19, with a special emphasis on the potential for these drugs to cause metabolism-dependent inhibition of CYP2C19. The implications of our results were explored by dynamic simulations assessing the level of active CYP2C19 under multiple doses of omeprazole. This physiologically based pharmacokinetic modeling and simulation provided additional insight into the ongoing debate surrounding the interaction between PPIs and clopidogrel.
Structures of the PPIs and omeprazole metabolites examined.
Materials and Methods
Chemicals and Reagents.
Omeprazole and 4′-hydroxymephenytoin were purchased from Sigma-Aldrich (St. Louis, MO). Esomeprazole, 5′-hydroxyomeprazole, S-mephenytoin, lansoprazole, omeprazole sulfide, and pantoprazole were purchased from Toronto Research Chemicals (North York, ON, Canada). R-Omeprazole and omeprazole sulfone were purchased from SynFine Research (Richmond Hill, ON, Canada) or Toronto Research Chemicals. Human liver microsomes (pooled from 16 donors) and human hepatocytes were prepared from nontransplantable livers and characterized at XenoTech, LLC (Lenexa, KS) as described previously (Parkinson et al., 2011), and recombinant CYP2C19 (Bactosomes) was obtained from Cypex (Dundee, Scotland). All other reagents were obtained from commercial sources, as detailed elsewhere (Ogilvie et al., 2006; Nassar et al., 2009; Parkinson et al., 2011).
CYP2C19 Inhibition: IC50 Determinations.
CYP2C19 activity in HLM was determined according to previously published procedures (Ogilvie et al., 2006; Nassar et al., 2009; Parkinson et al., 2011). In brief, incubations were conducted in 200-μl incubation mixtures (pH 7.4) containing high-purity water, potassium phosphate buffer (50 mM), MgCl2 (3 mM), EDTA (1 mM), NADP (1 mM), glucose 6-phosphate (5 mM), glucose-6-phosphate dehydrogenase (1 unit/ml), S-mephenytoin (approximately equal to Km, i.e., 40 μM, final), and HLM (0.1 mg protein/ml, except where otherwise noted). All incubations were conducted in duplicate at 37°C for 5 min (except where otherwise noted) and were terminated by the addition of 200 μl of acetonitrile containing an internal standard (d3-4′-hydroxymephenytoin). Aliquots of the stock and/or working solutions of the inhibitors (i.e., omeprazole, esomeprazole, R-omeprazole, lansoprazole, or pantoprazole; final concentrations ranging from 0.1 to 100 μM in methanol) were added to buffer mixtures containing the components described above but before addition of the NADPH-generating system. To evaluate the potential for metabolism-dependent inhibition, the inhibitors (at the same concentrations used to evaluate direct inhibition) were incubated at 37°C with NADPH-fortified HLM for approximately 30 min. After the 30-min preincubation, S-mephenytoin (40 μM, final) was added, and the incubation was continued for 5 min to measure residual P450 activity. Precipitated protein was pelleted by centrifugation (920g for 10 min at 10°C). Calibration and quality control standards (4′-hydroxymephenytoin) were prepared in zero-time incubations. IC50 determinations with recombinant human CYP2C19 (15 pmol/ml) were conducted in the same manner except that the incubation time was only 2 min to prevent overmetabolism of omeprazole. Metabolite formation (4′-hydroxymephenytoin) was analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described previously (Ogilvie et al., 2006; Parkinson et al., 2011).
Incubations with cryopreserved human hepatocytes (pooled, n = 3, 106 cells/ml) were conducted in 200-μl incubation mixtures at approximately 37°C in Krebs Henseleit buffer in triplicate. In all cases, the solvent or omeprazole was allowed to equilibrate for 10 min with hepatocytes before incubations. For samples with no preincubation with inhibitor, reactions were started by the addition of hepatocytes to prewarmed Krebs Henseleit buffer containing inhibitor and S-mephenytoin (40 μM, final concentration). For reactions with a preincubation, hepatocytes and inhibitor were incubated at 37°C for 30 min, and reactions were started by addition of S-mephenytoin (40 μM, final). In all cases, marker substrate reactions were conducted for 60 min and terminated by the addition of an equal volume of acetonitrile and internal standard (d3-4′-hydroxymephenytoin). Metabolite formation (4′-hydroxymephenytoin) was analyzed by LC-MS/MS as described previously (Ogilvie et al., 2006; Parkinson et al., 2011).
Metabolic Stability of Omeprazole.
The metabolic stability of omeprazole (10 μM) was determined at three concentrations of HLM (0.1, 1, and 2.5 mg/ml) under conditions similar to those described under CYP2C19 Inhibition: IC50 Determinations for CYP2C19 inhibition experiments. Omeprazole was incubated for 0, 5, 10, 20, 30, 45, and 60 min in triplicate. Reactions were terminated by the addition of an equal volume of acetonitrile and internal standard (pantoprazole). Precipitated protein was removed by centrifugation (920g for 10 min at 10°C). Omeprazole disappearance was monitored by LC-MS/MS. Calibration standards were prepared in zero-time incubations.
Microsomal Binding of Omeprazole.
The binding of omeprazole to microsomal protein was determined by ultrafiltration with Millipore Amicon Centriplus centrifugal filter devices (15 ml, 30-kDa membrane) obtained from Thermo Fisher Scientific (Waltham, MA). Omeprazole (2 and 10 μM), was incubated with pooled HLM (0, 0.1, 1, or 2.5 mg/ml) as described under Metabolic Stability of Omeprazole, but in the absence of an NADPH-generating system, at 37°C for 10 min. Aliquots (1.1 ml) were then removed and added to the ultrafiltration devices and centrifuged at 1900g in a Sorvall (Asheville, NC) RC 5C centrifuge with a Sorvall SS-34 rotor at room temperature for 5 min. Aliquots of the ultrafiltrate (100 μl) were transferred to glass tubes, and an equal volume of acetonitrile was added and vortexed. Precipitated protein was removed by centrifugation (920g for 10 min at 10°C). After centrifugation, an aliquot (100 μl) was transferred to an equal volume of acetonitrile (with pantoprazole as the internal standard) and analyzed for omeprazole concentration by LC-MS/MS.
KI and kinact Determinations.
To determine the KI and kinact values for the inactivation of CYP2C19, various concentrations of omeprazole (1–60 μM) were incubated for 2.5 to 15 min with pooled human liver microsomes (0.1 and 2.5 mg/ml) at 37°C. For experiments conducted at 0.1 mg/ml HLM, after the preincubations, S-mephenytoin (40 or 400 μM final concentration) was added and residual CYP2C19 activity was determined as described under CYP2C19 Inhibition: IC50 Determinations. For experiments conducted at 2.5 mg/ml, after the preincubation, an aliquot (8 μl) was transferred to another incubation tube (final volume 200 μl) containing S-mephenytoin (400 μM) and an NADPH-generating system to measure residual CYP2C19 activity as described under CYP2C19 Inhibition: IC50 Determinations. This procedure diluted the microsomes to 0.1 mg/ml and diluted omeprazole to 1/25th of its original concentration. Reactions were performed in triplicate.
CYP2C19 Activity in Cultured Human Hepatocytes.
Cultured human hepatocytes were prepared and treated, and microsomes (0.02 mg/ml) were isolated as described previously (Nassar et al., 2009). In brief, cultured human hepatocytes were treated for 72 h with 0.1% (v/v) dimethyl sulfoxide (DMSO) (control) or 100 μM omeprazole. Isolated microsomes were washed and incubated for 30 min with marker substrate (40 μM S-mephenytoin) to determine CYP2C19 activity as described under CYP2C19 Inhibition: IC50 Determinations.
Assessment of MDI Reversibility by Ultracentrifugation.
Omeprazole (100 μM), R-omeprazole (100 μM), and esomeprazole (100 μM) were incubated in triplicate with NADPH-fortified pooled HLM (0.1 mg/ml) at 37°C for 30 min in potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM, pH 7.4), and chemically reduced NADPH (1 mM) as described previously (Parkinson et al., 2011). Incubations (n = 3) with solvent alone (1% methanol v/v, final) served as controls. After the 30-min incubation, HLM were 1) assayed directly for residual CYP2C19 activity; 2) re-isolated by ultracentrifugation and then assayed for residual CYP2C19 activity; or 3) treated with potassium ferricyanide, re-isolated by ultracentrifugation, and then assayed for residual CYP2C19 activity. For samples in groups 2 and 3, microsomal protein was re-isolated by ultracentrifugation (100,000g for 30 min at 4°C in a Beckman ultracentrifuge with a 70 Ti rotor; Beckman Coulter, Fullerton, CA). The supernatant fraction was discarded, and the resultant microsomal pellets were rinsed three times with wash buffer (150 mM potassium chloride and 10 mM EDTA, pH 7.4) to remove residual omeprazole and/or any reversible inhibitory metabolites. Microsomal pellets were resuspended in 250 mM sucrose, and the microsomal protein concentration was determined by the Pierce BCA protein assay (Thermo Fisher Scientific). For samples in group 3, HLM were incubated with potassium ferricyanide (2 mM) for 10 min at 37°C before re-isolation of microsomal protein by ultracentrifugation to disrupt nitrogen-based metabolite inhibitory complexes (Franklin, 1977). Residual CYP2C19 activity was assessed at a final concentration of 0.1 mg/ml HLM (supplemented with an NADPH-regenerating system) with S-mephenytoin (400 μM, i.e., 10 × Km) to reduce the inhibitory effects of any residual competitive inhibition.
Analytical Methods.
LC-MS/MS methods were performed as described previously (Ogilvie et al., 2006; Parkinson et al., 2011). Omeprazole analysis was performed with an Applied Biosystems/Sciex API2000 high-performance liquid chromatography-tandem mass spectroscopy system equipped with an electrospray (TurboIonSpray) ionization source (Applied Biosystems, Foster City, CA), two LC-10ADvp pumps with an SCL-10Advp controller, a SIL-HTA autosampler, and a DGU-14 solvent degasser (Shimadzu, Kyoto, Japan). The high-performance liquid chromatography column used was an Atlantis dC18, 5 μm, 100- by 2-mm column (Waters, Milford, MA), which was preceded by a direct connection guard column with a C8, 4- by 2-mm cartridge (Waters). Masses were monitored in the multiple reaction monitoring mode: omeprazole 345.9 → 197.9 amu and internal standard, pantoprazole, 383.9 → 199.9 amu. Mobile phases were buffer A (0.2% formic acid in water) and buffer B (0.2% formic acid in methanol). Omeprazole and pantoprazole were eluted with a linear gradient (25% B to 75% B) over 2.5 min.
Data Analyses and Simulations.
All IC50, half-life, KI, and kinact values were determined by nonlinear regression with XLfit3 (version 3.0.5; ID Business Solutions Ltd., Guildford, Surrey, UK), which is an add-in for the computer program Microsoft Excel (Office 2003; Microsoft, Redmond, WA) or with GraFit (version 4.0.21; Erithacus Software, Horley, Surrey, UK), as detailed previously (Ogilvie et al., 2006; Nassar et al., 2009; Parkinson et al., 2011).
In vitro-to-in vivo extrapolation of CYP2C19 inactivation was performed using a mechanistic static model (MSM) (Grimm et al., 2009) and a mechanistic dynamic model (MDM) (Rowland Yeo et al., 2010). The MSM was implemented based on the following equation:
 where fm,CYP represents the fraction of a hypothetical concomitantly administered drug metabolized by a given P450 enzyme, [I] is the inactivator concentration, kinact and KI are the in vitro inactivation parameters, and kdeg is the rate constant for enzyme degradation [which has not been determined experimentally in vivo for CYP2C19, but for which the average value was reported to be 0.000445 min−1 on the basis of in vitro data (Renwick et al., 2000; Yang et al., 2008)]. Given that the unbound plasma Cmax was previously found to be more predictive than the total Cmax for MDIs (Obach et al., 2007; Grimm et al., 2009), this value was used in the MSM (eq. 1) and was based on the total omeprazole plasma Cmax of 3.87 μM after 5 days of dosing (40 mg daily) in CYP2C19-extensive metabolizers (EMs) (Hassan-Alin et al., 2005), coupled with its plasma protein binding of 95%, for an unbound plasma Cmax of 0.19 μM.
where fm,CYP represents the fraction of a hypothetical concomitantly administered drug metabolized by a given P450 enzyme, [I] is the inactivator concentration, kinact and KI are the in vitro inactivation parameters, and kdeg is the rate constant for enzyme degradation [which has not been determined experimentally in vivo for CYP2C19, but for which the average value was reported to be 0.000445 min−1 on the basis of in vitro data (Renwick et al., 2000; Yang et al., 2008)]. Given that the unbound plasma Cmax was previously found to be more predictive than the total Cmax for MDIs (Obach et al., 2007; Grimm et al., 2009), this value was used in the MSM (eq. 1) and was based on the total omeprazole plasma Cmax of 3.87 μM after 5 days of dosing (40 mg daily) in CYP2C19-extensive metabolizers (EMs) (Hassan-Alin et al., 2005), coupled with its plasma protein binding of 95%, for an unbound plasma Cmax of 0.19 μM.
The MDM simulations were performed using the Simcyp Population-Based Simulator (version 10.2; Simcyp Limited, Sheffield, UK). The differential equations that form the basis of these simulations are described in detail elsewhere (Rowland Yeo et al., 2010). Input parameters can be found in the supplemental data.
The purpose of the simulations was to assess the time-varying effect of repeated omeprazole administration (40 mg every 12 h) on the level of active CYP2C19 in liver. This included the self-inhibition effect because the deactivation of CYP2C19 led to lower clearance and higher concentrations of omeprazole itself. The simulations were performed twice using the lower (1.7 μM) and upper (9.1 μM) boundaries of observed KI values and taking into account an unbound fraction of 0.75 (i.e., the free fraction of omeprazole in HLM at 1–2.5 mg/ml). kinact was assumed to be 0.045 min−1, and turnover of CYP2C19 was the same as that assumed for the MSM.
Although the main purpose of the simulations was to assess the level of active enzyme, S-mephenytoin was used as a substrate on day 14 after administration of omeprazole, and the effect on AUC was simulated on day 7 after administration of omeprazole. The omeprazole dose was continued until day 14, and the AUC for S-mephenytoin was assessed from day 7 until the end of the study (day 14) and compared with AUC in the absence of an inactivator.
Results
Inhibitory Effect of Omeprazole on S-Mephenytoin 4′-Hydroxylation in Multiple Test Systems: IC50 Determinations, Metabolic Stability, and Microsomal Protein Binding.
Omeprazole was evaluated as a direct-acting and MDI of CYP2C19 activity (S-mephenytoin 4′-hydroxylation) in pooled human liver microsomes (n = 16), recombinant CYP2C19 (Bactosomes), and pooled, cryopreserved human hepatocytes (n = 3) at a substrate concentration approximately equal to Km (40 μM). The results are summarized in Fig. 2, a to d, and Table 1. The results show that omeprazole caused metabolism-dependent inhibition of CYP2C19 as evidenced by a left shift in the IC50 curves after a 30-min preincubation with NADPH-fortified HLM, recombinant CYP2C19, and human hepatocytes (IC50 shifts of 4.1-, 6.9-, and 3.6-fold, respectively). A left shift of ≥1.5-fold is considered indicative of metabolism-dependent inhibition (Parkinson et al., 2011). With no preincubation, omeprazole inhibited CYP2C19 in all three systems with similar IC50 values ranging from 4.7 to 9.6 μM, which decreased to approximately 1.5 μM in all three test systems after preincubation with NADPH. The experiment presented in Fig. 2a also included an IC50 determination with a 30-min preincubation step without NADPH to confirm that the IC50 shift was dependent on NADPH (Table 1; for clarity, these data are not presented in Fig. 2a). Omeprazole was also examined as an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP3A4, and CYP2J2 and was found to be a weak direct inhibitor of these enzymes (IC50 > approximately 100 μM), with no NADPH-dependent IC50 shift > 1.5-fold (see supplemental data).
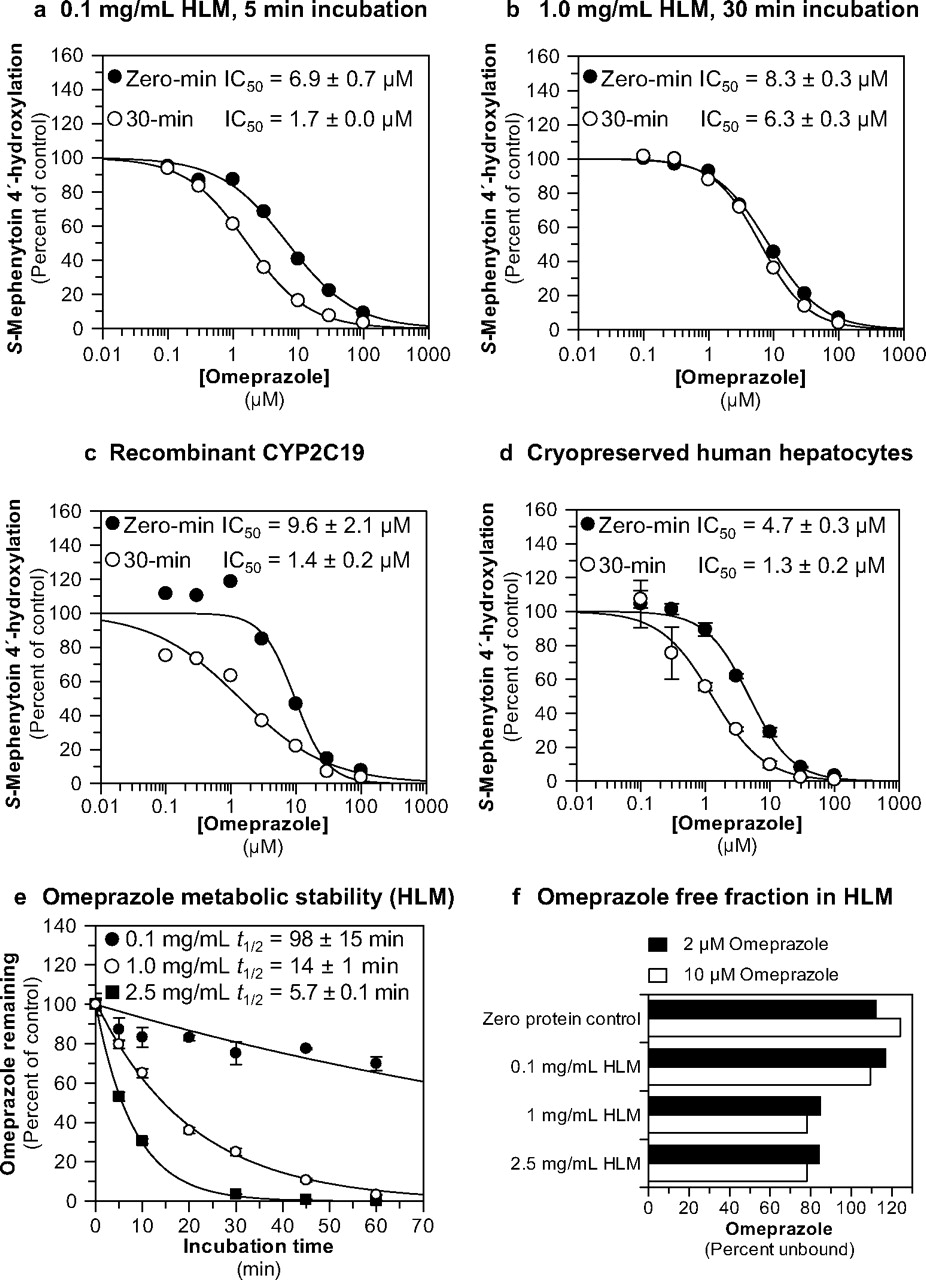
Evaluation of omeprazole as a direct-acting and MDI of CYP2C19. Each symbol represents the average of duplicate determinations unless otherwise indicated. a, omeprazole inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 97.3 and 95 pmol · mg−1 · min−1 with and without preincubation, respectively). b, omeprazole inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 97.3 and 74.3 pmol · mg−1 · min−1 with and without preincubation, respectively). c, omeprazole inhibited recombinant human CYP2C19 with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 0.690 and 0.828 min−1 with and without preincubation, respectively). d, omeprazole inhibited CYP2C19 cryopreserved human hepatocytes with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 24.7 and 24.8 pmol/million cells/min with and without preincubation, respectively). Each symbol represents the average of triplicate determinations, and error bars represent the S.D. e, the metabolic stability of omeprazole (10 μM) was evaluated in HLM at the protein concentrations used in this study (0.1, 1, and 2.5 mg/ml), as described under Materials and Methods. Half-life values for the disappearance of omeprazole are as indicated. Each symbol represents the average of triplicate determinations, and error bars represent the S.D. f, the microsomal binding of omeprazole (2 and 10 μM) was evaluated as described under Materials and Methods.
Inhibition of CYP2C19 in human liver microsomes by omeprazole, its enantiomers, or its major metabolites and lansoprazole and pantoprazole
The data presented in Fig. 2a were obtained under “low microsomal protein-short incubation time” conditions (i.e., 0.1 mg/ml HLM for 5 min). However, S-mephenytoin is a low-turnover substrate in HLM, for which reason S-mephenytoin 4′-hydroxylation activity is frequently assessed by incubating S-mephenytoin with high concentrations of HLM [i.e., ≥0.2 mg/ml) for 20 to 40 min (e.g., Walsky and Obach, 2004; Nishiya et al., 2009; Parkinson et al., 2011)]. Because metabolism-dependent inhibition of CYP2C19 by omeprazole had not been previously described, we also examined the ability of omeprazole to inhibit CYP2C19 under “high microsomal protein-long incubation time” conditions (i.e., 1 mg/ml HLM for 30 min). Under such conditions we detected no metabolism-dependent inhibition of CYP2C19 by omeprazole (i.e., IC50 shift <1.5; Fig. 2b).
Because of the difference in results between low microsomal protein-short incubation time and high microsomal protein-long incubation time conditions, we examined the metabolic stability and nonspecific binding of omeprazole in HLM. In NADPH-fortified HLM at 0.1 mg/ml, omeprazole was relatively stable with a half-life of 98 min (Fig. 2e). However, at 1 and 2.5 mg/ml HLM (i.e., the concentration used in one of the kinact determinations, see Inhibitory Effects of the Major Omeprazole Metabolites and Enantiomers on S-Mephenytoin 4′-Hydroxylation in Human Liver Microsomes: IC50 Determinations) omeprazole rapidly disappeared from incubations, with half-life values of only 14 and 5.7 min, respectively. Nonspecific binding of omeprazole to HLM was also examined (by ultrafiltration) as a possible cause of the discrepancy in results between low microsomal protein-short incubation time and high microsomal protein-long incubation time conditions. However, as shown in Fig. 2f, >75% of omeprazole (2 and 10 μM) remained free in the incubation from 0.1 to 2.5 mg/ml.
Inhibitory Effects of the Major Omeprazole Metabolites and Enantiomers on S-Mephenytoin 4′-Hydroxylation in Human Liver Microsomes: IC50 Determinations.
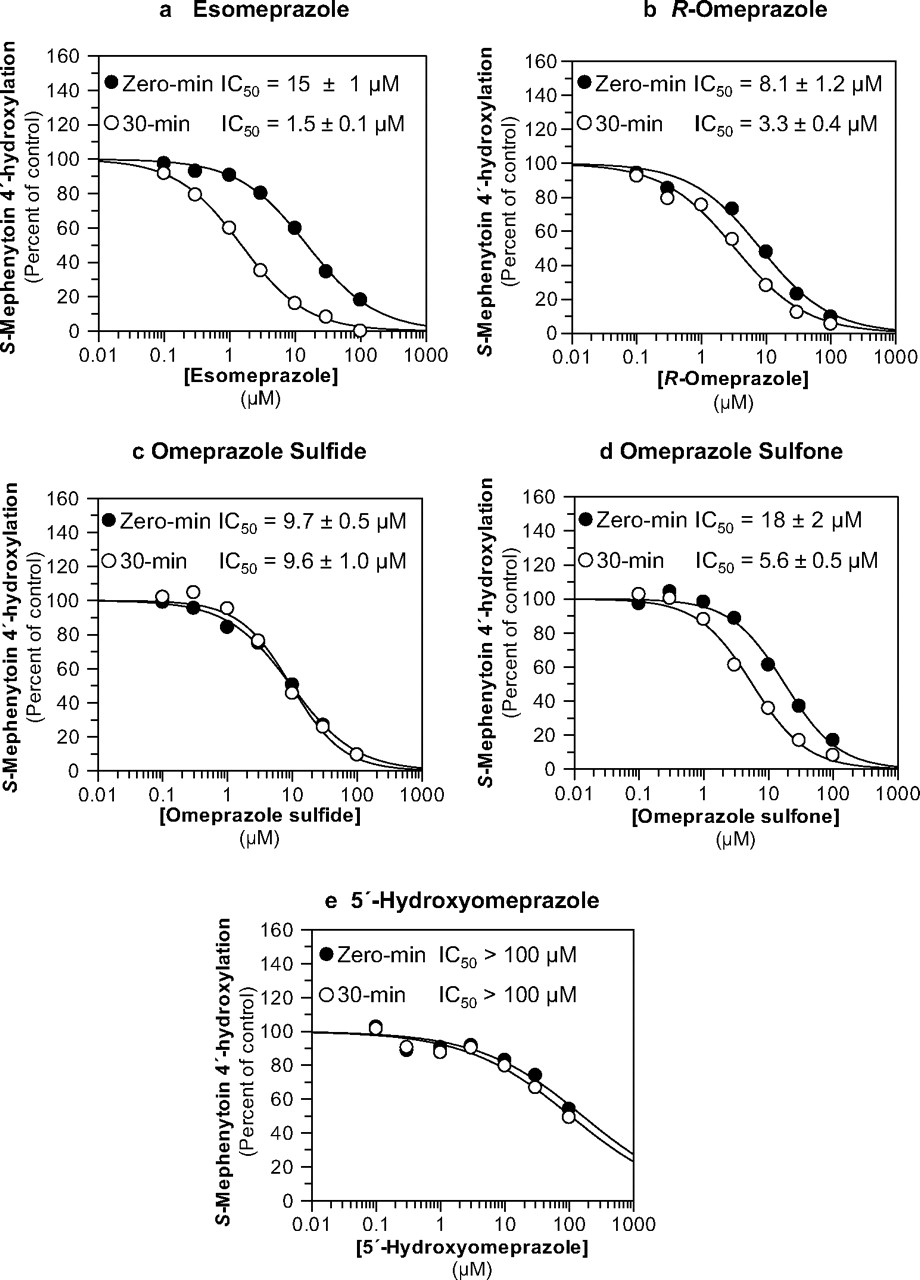
Esomeprazole, R-omeprazole, omeprazole sulfide, omeprazole sulfone, and 5′-hydroxyomeprazole were evaluated as direct-acting and MDIs of CYP2C19 activity (S-mephenytoin 4′-hydroxylation) in pooled human liver microsomes (n = 16, 0.1 mg/ml) at a substrate concentration approximately equal to the Km (40 μM). The results are summarized in Table 1 and Fig. 3. The results show that esomeprazole (Fig. 3a), R-omeprazole (Fig. 3b), and omeprazole sulfone (Fig. 3d) caused metabolism-dependent inhibition of CYP2C19 as evidenced by a left shift in the IC50 curves (>1.5-fold) after a 30-min preincubation with NADPH-fortified HLM (10-, 2.5-, and 3.2-fold IC50 shifts, respectively), with similar “shifted” IC50 values ranging from 1.5 to 5.6 μM. The IC50 values after a 30-min preincubation in the absence of NADPH were higher than those in the presence of NADPH, suggesting that the time-dependent inhibition of CYP2C19 by these compounds was in fact dependent on metabolism (Table 1). The IC50 values for omeprazole sulfide after a 30-min preincubation with or without NADPH remained similar to the value without preincubation (IC50 shift < 1.5-fold), suggesting that omeprazole sulfide is only a direct-acting inhibitor of CYP2C19 (Fig. 3c; Table 1). 5′-Hydroxyomeprazole, one of the major metabolites formed by CYP2C19, was a weak inhibitor of CYP2C19 (IC50 > 100 μM) (Fig. 3e). The experiments presented in Fig. 3 also included an IC50 determination with a 30-min preincubation step without NADPH to confirm that the IC50 shift was dependent on NADPH (Table 1; for clarity, these data are not presented in Fig. 3).

Evaluation of esomeprazole, R-omeprazole, omeprazole sulfide, omeprazole sulfone, and 5′-hydroxyomeprazole as direct-acting and MDIs of CYP2C19. Each symbol represents the average of duplicate determinations unless otherwise indicated. a, esomeprazole inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 80.9 and 88.1 pmol · mg−1 · min−1 with and without preincubation, respectively). b, R-omeprazole inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 106 and 108 pmol · mg−1 · min−1 with and without preincubation, respectively). c, omeprazole sulfide inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 89.8 and 82.2 pmol · mg−1 · min−1 with and without preincubation, respectively). d, omeprazole sulfone inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 85.4 and 84.7 pmol · mg−1 · min−1 with and without preincubation, respectively). e, 5′-hydroxyomeprazole weakly inhibited CYP2C19 in HLM (S-mephenytoin 4′-hydroxylation control rates = 108 and 97 pmol · mg−1 · min−1 with and without preincubation, respectively).
Inhibitory Effects of Lansoprazole and Pantoprazole on S-Mephenytoin 4′-Hydroxylation in Human Liver Microsomes and Cryopreserved Human Hepatocytes: IC50 Determinations.
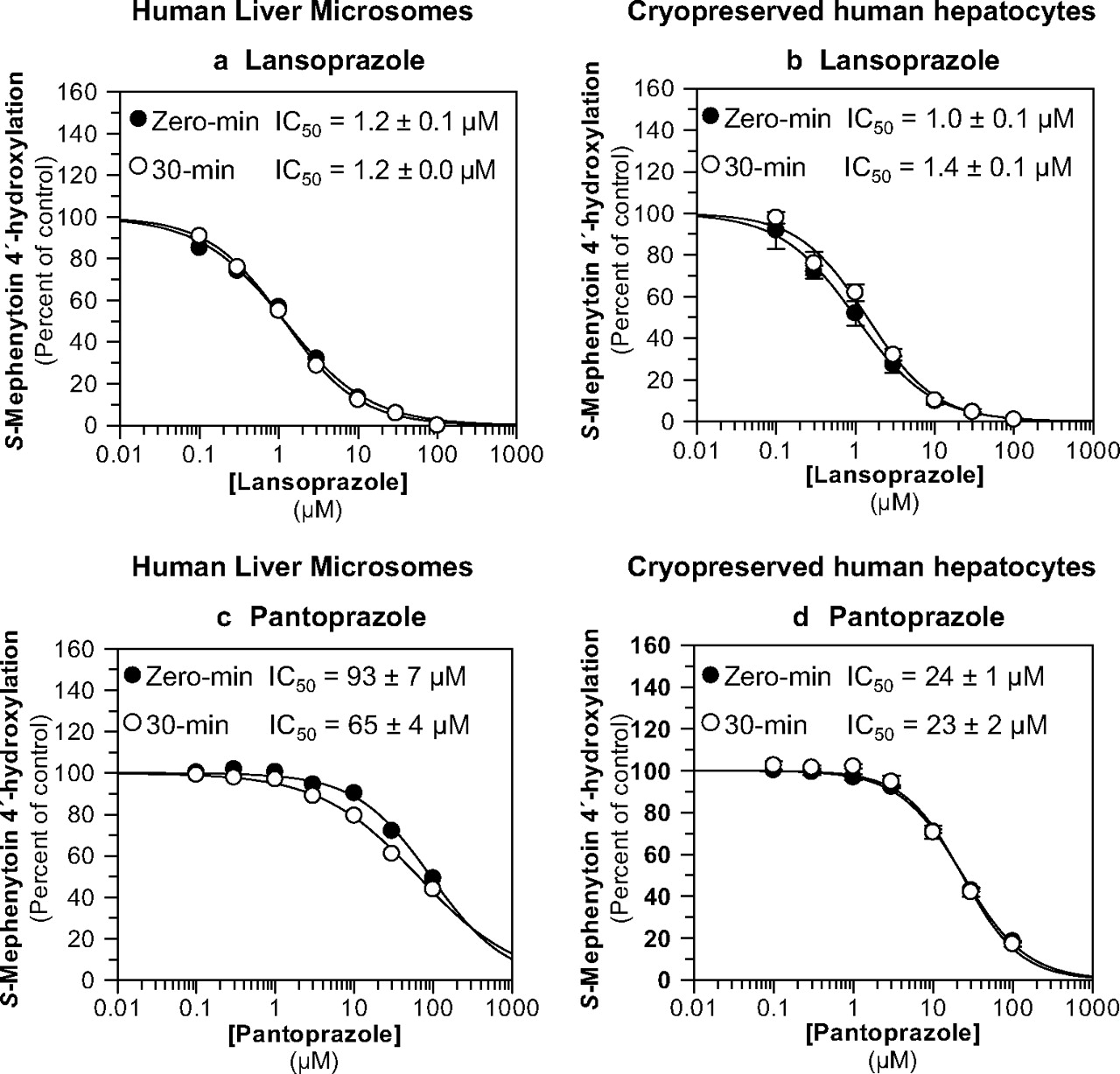
Lansoprazole and pantoprazole were evaluated as direct-acting and MDIs of CYP2C19 activity (S-mephenytoin 4′-hydroxylation) in pooled human liver microsomes (n = 16) and pooled, cryopreserved human hepatocytes (n = 3) at a substrate concentration approximately equal to the Km (40 μM). The results are summarized in Table 1 and Fig. 4. Neither lansoprazole nor pantoprazole caused metabolism-dependent inhibition of CYP2C19 in either HLM or cryopreserved human hepatocytes (i.e., IC50 shifts <1.5-fold). Lansoprazole was a relatively potent (i.e., IC50 ≈ 1 μM), direct-acting inhibitor of CYP2C19 in HLM and cryopreserved human hepatocytes (Fig. 4, a and b), whereas pantoprazole was a relatively weak inhibitor (Fig. 4, c and d). The experiments presented in Fig. 4, a and c, also included an IC50 determination with a 30-min preincubation step without NADPH to confirm that the IC50 shift was dependent on NADPH (Table 1; for clarity, these data are not presented in Fig. 4).
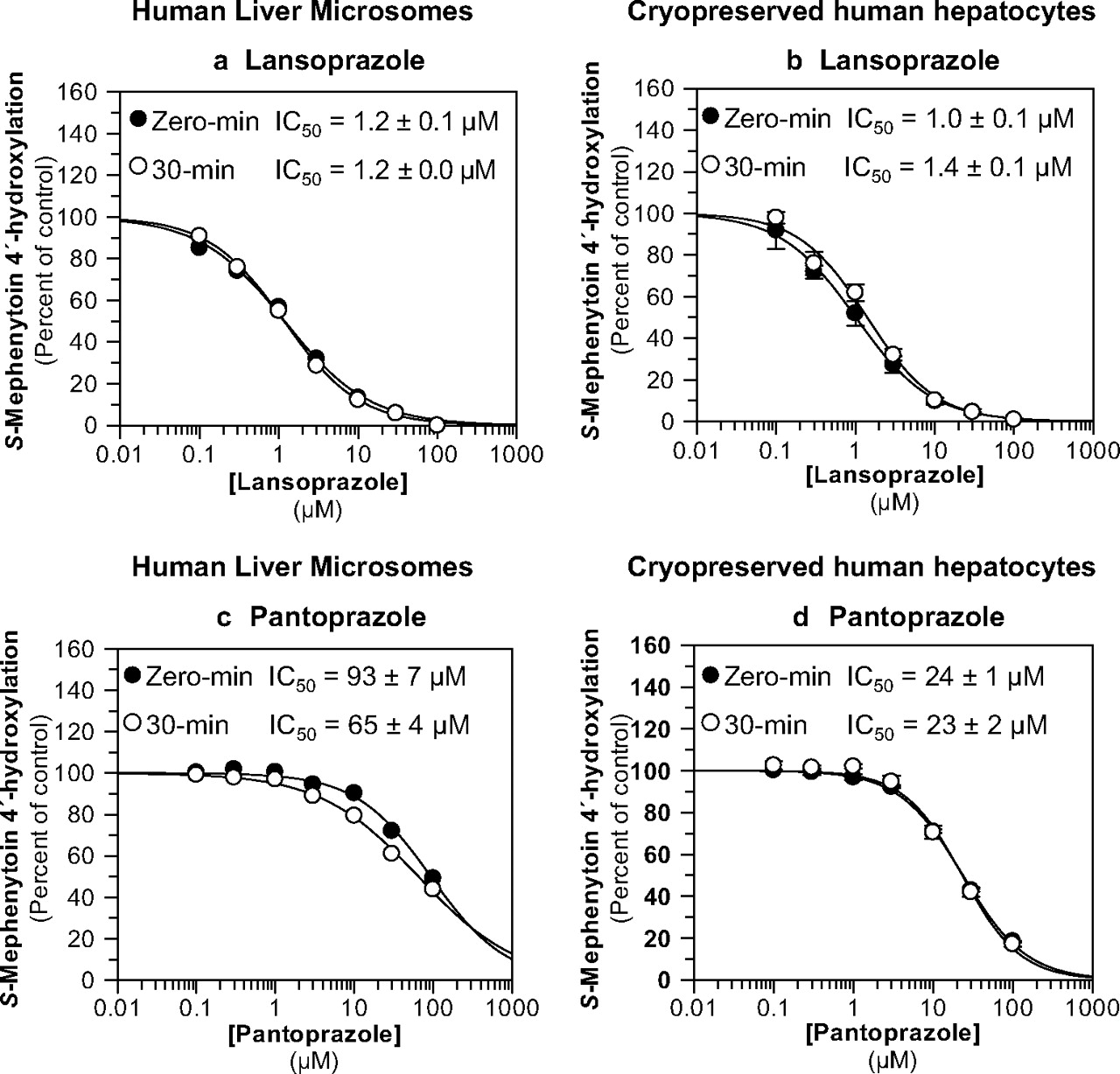
Evaluation of lansoprazole and pantoprazole as direct-acting and MDIs of CYP2C19. Each symbol represents the average of duplicate determinations unless otherwise indicated. a, lansoprazole inhibited CYP2C19 in HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 83.8 and 79.6 pmol · mg−1 · min−1 with and without preincubation, respectively). b, lansoprazole inhibited CYP2C19 in cryopreserved human hepatocytes with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 30.4 and 26.6 pmol/million cells/min with and without preincubation, respectively). Each symbol represents the average of triplicate determinations, and error bars represent the S.D. c, pantoprazole inhibited CYP2C19 in pooled HLM with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 61.7 and 71 pmol · mg−1 · min−1 with and without preincubation, respectively). d, pantoprazole inhibited CYP2C19 in cryopreserved human hepatocytes with IC50 values as shown (S-mephenytoin 4′-hydroxylation control rates = 24.5 and 14.2 pmol/million cells/min with and without preincubation, respectively). Each symbol represents the average of triplicate determinations, and error bars represent the S.D.
Inactivation of CYP2C19 by Omeprazole: KI and kinact Determinations.
The results in Fig. 2 suggest that omeprazole is an MDI of CYP2C19. Accordingly, experiments were performed to determine KI (the concentration of omeprazole supporting half-maximal rate of CYP2C19 inactivation) and kinact (the first-order rate constant for CYP2C19 inactivation). The results from three different experiments are summarized in Fig. 5. The three experimental conditions were 1) low HLM concentration with [S] = Km with no dilution step (Fig. 5, a and b), 2) low HLM concentration with [S] = 10 × Km (Fig. 5, c and d), and 3) high [HLM] with [S] = 10 × Km with a 25-fold dilution step (Fig. 5, e and f). Under all three conditions, the inactivation of CYP2C19 was dependent on the concentration of omeprazole (over the full ranges examined), and the time course conformed to a first-order inactivation process (as indicated by the linearity of plots of the log of the residual enzyme activity against time; Fig. 5, a, c, and e). The KI values were different depending on whether omeprazole was preincubated with human liver microsomes at 2.5 mg/ml (and subsequently diluted 25-fold to determine residual CYP2C19 activity with substrate = 10 Km; Fig. 5, e and f) or 0.1 mg/ml (and not diluted to determine residual CYP2C19 activity, with substrate = 10 Km or Km; Fig. 5, a–d). The KI value was approximately 9 μM in the former case and approximately 2 μM in the latter cases. In all three cases, the kinact values were approximately 0.04 min−1, which means that, in the presence of saturating concentrations of omeprazole, 4% of CYP2C19 was inactivated every minute. The efficiency of CYP2C19 inactivation (kinact/KI) decreased by a factor of approximately 5 when the concentration of HLM was increased and a dilution step was used. Taken together, these data suggest that omeprazole is an irreversible inactivator of CYP2C19.

Determination of KI and kinact for the metabolism-dependent inhibition of CYP2C19 by omeprazole. Individual points represent the average of triplicate determinations ±S.D. unless otherwise noted. For graphs in a and c, omeprazole was preincubated (at concentrations indicated in the legend of c), and residual CYP2C19 activity was determined as described under Materials and Methods. For the graph in e, omeprazole was preincubated (at concentrations indicated), and residual CYP2C19 activity was determined as described under Materials and Methods (after a 25-fold dilution). The graphs in b, d, and f represent the direct plots of the initial rates of inactivation of CYP2C19. Values are the slopes of the initial rates of inactivation (kobs) at each concentration of omeprazole (shown ±S.E.).
Effects of Omeprazole on CYP2C19 Activity in Microsomes Prepared from Hepatocytes after 3 Days of Treatment.
CYP2C19 activity (S-mephenytoin 4′-hydroxylation) was measured in microsomes isolated from fresh primary cultures of human hepatocytes treated with omeprazole (100 μM; 3 days). It should be noted that this concentration is approximately 19 times greater than the Cmax in poor metabolizers (PMs) [5.3 μM (Chen et al., 2009); who can be neither induced nor inhibited with respect to CYP2C19] and 26 times greater than the Cmax in EMs (Hassan-Alin et al., 2005) after a 40-mg oral dose of omeprazole. Omeprazole treatment decreased microsomal CYP2C19 activity in hepatocytes that initially expressed high levels of CYP2C19, but it increased microsomal CYP2C19 activity in hepatocytes that initially expressed low levels of CYP2C19 (Fig. 6). This dual effect likely reflects the overall effect of two opposing actions of omeprazole; in hepatocytes with high CYP2C19 activity, the predominant effect of omeprazole was irreversible inactivation, but in hepatocytes with low CYP2C19 activity, the predominant effect was induction via pregnane X receptor activation (Yueh et al., 2005). These data were obtained from many studies conducted to examine the potential for drug candidates to induce P450 enzymes, in which primary cultures of human hepatocytes were treated with 100 μM omeprazole, a well known aryl hydrocarbon receptor activator and therefore a positive control for CYP1A2 induction. Because the microsomes are obtained from these studies in a way that washes out residual inhibitor, these data provided additional evidence that omeprazole is an irreversible inactivator of CYP2C19.
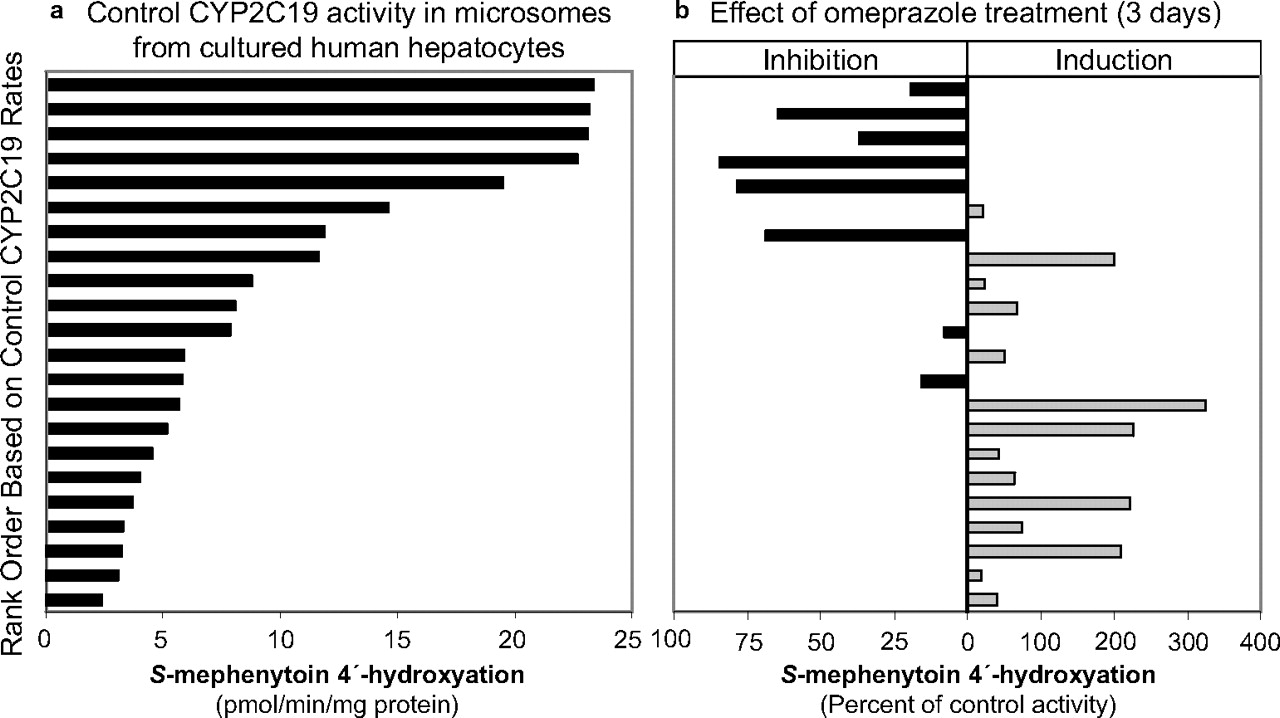
CYP2C19 activity (S-mephenytoin 4′-hydroxylation) in microsomes isolated from fresh-plated hepatocytes treated with DMSO (control) or omeprazole. Primary cultures of human hepatocytes were treated for 3 consecutive days with DMSO or omeprazole (100 μM), and microsomes were prepared from the hepatocytes 24 h after the last treatment. CYP2C19 activity was determined as described under Materials and Methods. Activities were sorted in rank order from highest to lowest control rates (a), and the corresponding omeprazole-treated samples are displayed in b in terms of percentage of control activity.
Irreversible or Quasi-Irreversible Inhibition of CYP2C19 by Omeprazole and Esomeprazole, but Not R-omeprazole: Ultracentrifugation.
Because esomeprazole was approximately 4-fold more effective as a MDI of CYP2C19 than R-omeprazole (Table 1; Fig. 3, a and b) with a 10-fold shift in IC50 value for the former and a 2.5-fold shift for the latter, omeprazole and its individual enantiomers were further examined to determine whether the inactivation of CYP2C19 involved irreversible or quasi-irreversible inhibition on the basis of an ultracentrifugation method described herein and previously (Parkinson et al., 2011). A concentration of inhibitor that caused nearly complete (i.e., >∼95%) inhibition after 30-min preincubation with NADPH, but incomplete (<∼85%) inhibition after 30-min preincubation in the absence of NADPH was chosen (i.e., 100 μM for all). Pooled HLM were treated with inhibitor or solvent (methanol, 0.1% v/v) for 30 min in the presence of NADPH and in the presence or absence of potassium ferricyanide (to reverse the formation of metabolite inhibitory complex associated with quasi-irreversible inactivation) (Fig. 7). After the 30-min incubation, pooled HLM samples were either 1) assayed directly for residual P450 activity (“prespin”); 2) re-isolated by ultracentrifugation and then assayed for residual P450 activity (“postspin”); or 3) treated with potassium ferricyanide, re-isolated by ultracentrifugation, and then assayed for residual P450 activity (“postspin + K3[Fe(CN)6]”). As expected, substantial inhibition was observed after treatment with omeprazole, esomeprazole, and R-omeprazole (Fig. 7, prespin) in a rank-order consistent with the IC50 shifts: esomeprazole ≈ omeprazole > R-omeprazole. It was surprising to note that centrifugation and re-isolation of HLM after preincubation largely reversed the inhibition caused by R-omeprazole (Fig. 7, postspin and postspin + K3[Fe(CN)6]). In contrast, centrifugation did not fully restore CYP2C19 activity after treatment with omeprazole or esomeprazole, which suggests that esomeprazole is the major contributor to the inactivation of CYP2C19 by omeprazole. Potassium ferricyanide treatment of the samples caused some additional restoration of activity after treatment with omeprazole and esomeprazole, but it did not fully restore CYP2C19 activity. Taken together, these results suggest that the metabolism-dependent inhibition of CYP2C19 observed with racemic omeprazole is largely irreversible (because of covalent binding to the apo protein, heme moiety, or both). However, given the partial restoration of CYP2C19 activity with potassium ferricyanide, contribution of a quasi-irreversible mechanism (because of metabolite inhibitory complex formation) cannot be completely ruled out. The results of a previous experiment that was based on overnight dialysis are also consistent with an irreversible or quasi-irreversible mechanism of the metabolism-dependent inhibition of CYP2C19 with racemic omeprazole (Paris et al., 2008). However, the results presented in Fig. 7 show that the metabolism-dependent inhibition of CYP2C19 caused by R-omeprazole is largely reversible after re-isolation of HLM, suggesting that one or more metabolites of this enantiomer is simply a more potent inhibitor of CYP2C19 than the parent, whereas metabolism of esomeprazole largely leads to irreversible inactivation of CYP2C19.

Reversibility assessment of the metabolism-dependent inhibition of CYP2C19 by omeprazole and its enantiomers with the ultracentrifugation method. The potential reversibility of the metabolism-dependent inhibition of CYP2C19 NADPH-fortified HLM (0.1 mg/ml) by omeprazole and its individual enantiomers (100 μM) was evaluated with the ultracentrifugation method as described under Materials and Methods. Incubations labeled “prespin” were conducted similarly to analogous samples in the IC50 determinations (conduced in triplicate and displayed as the average rates of S-mephenytoin 4′-hydroxylation ± S.D.). For incubations labeled postspin (conducted in triplicate and analyzed in triplicate), microsomal protein was isolated by ultracentrifugation after 30-min incubations with the inhibitor and NADPH and analyzed in triplicate for residual CYP2C19 activity (S-mephenytoin 4′-hydroxylation) displayed as the average rates ± S.E. as described under Materials and Methods. Half of the incubations from the postspin samples included potassium ferricyanide (2 mM, final concentration) as indicated. Samples treated with methanol (1% v/v, final) served as controls. All rates were normalized to final microsomal protein concentrations as described under Materials and Methods.
Simulation of Time-Dependent Changes in Active CYP2C19.
Active levels of hepatic CYP2C19 in the presence of omeprazole (40 mg twice daily for 14 days) were simulated with Simcyp version 10.2 as described under Materials and Methods. Figure 8 shows the active CYP2C19 levels with time. Depending on the assumption regarding KI (1.7 or 9.1 μM), the level of active enzyme could decrease to approximately 10 to 60% of the baseline, which would be associated with a 1.4- to 10-fold increase in the AUC of compounds mainly metabolized by CYP2C19. However, the simulated effect of omeprazole on the S-mephenytoin AUC showed only a 1.45-fold increase when the KI value was 9.1 μM and a 5.46-fold increase when the KI value was 1.7 μM, rather than a 10-fold increase, partly because of the contribution of other enzymes in its metabolism (e.g., CYP2B6) and possibly due to the inhibitory contribution of metabolites (e.g., omeprazole sulfone), not accounted for in the simulation.

Simulation of time-dependent changes in active CYP2C19. Active levels of hepatic CYP2C19 in the presence of omeprazole (40 mg twice daily for 14 days) were simulated with the Simcyp Simulator version 10.2, as described under Materials and Methods. a is based on a KI value of 1.7 μM; b is based on a KI value of 9.1 μM.
Discussion
Given the relatively few drugs that are metabolized extensively by CYP2C19, it is not surprising that few DDIs have been attributed to clinically relevant inhibition of CYP2C19. As noted in the Introduction, there is evidence of clinically relevant inhibition of CYP2C19 by omeprazole, but direct competitive inhibition of CYP2C19 is an unlikely cause; if it were, lansoprazole would be expected to inhibit CYP2C19 more than omeprazole, whereas the converse is observed clinically. It should be noted that the dose of lansoprazole versus omeprazole (which depends on indication) could partly explain this difference. For the lowest dose indications (i.e., gastroesophageal reflux disease or maintenance of healing of erosive esophagitis), it is 20 mg daily for omeprazole (Prilosec prescribing information, 2011, http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/019810s092,022056s008lbl.pdf) and 15 mg daily for lansoprazole (Prevacid prescribing information, 2010, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020406s074,021428s021lbl.pdf). For the highest dose indication (i.e., Zollinger-Ellison or other hypersecretory syndromes), the doses are equal (60 mg daily). On the basis of this comparison alone, it seems unlikely that the slight difference in the low dose explains the differences in clinical DDIs. This supposition is borne out by clinical data in the MTDI: negative interactions were reported between lansoprazole (60 mg daily for 10 days) and the CYP2C19 substrates diazepam and phenytoin (lansoprazole dose 60 mg daily for 9 days with the latter drug). No positive DDIs with CYP2C19 substrates have been reported in the MTDI with lansoprazole at any dose. On the other hand, with a low dose of omeprazole (20 mg daily) in various studies (8–23 days), the exposure (AUC) of the CYP2C19 substrates diazepam and escitalopram increased from 26 to 91%.
To our knowledge, with the exception of our preliminary findings (Paris et al., 2008; Parkinson et al., 2010), in vitro evidence for metabolism-dependent inhibition of CYP2C19 has not been previously described for any PPI. In the current study, omeprazole, esomeprazole, R-omeprazole, and omeprazole sulfone were identified as MDIs of CYP2C19 (IC50 shifts after a 30-min preincubation with NADPH of 4.2, 10, 2.5, and 3.2, respectively), whereas lansoprazole and pantoprazole were not MDIs (IC50 shifts <1.5). Furthermore, the metabolism-dependent inhibition of CYP2C19 by omeprazole and esomeprazole was not reversed by ultracentrifugation, suggesting that the inhibition was irreversible, whereas ultracentrifugation largely reversed such effects of R-omeprazole. Under various conditions, omeprazole inactivated CYP2C19 with KI values of 1.7 to 9.1 μM and kinact values (maximal rate of inactivation) of 0.041 to 0.046 min−1 [corresponding to kinact/KI values ranging from 5.1 to 24 min−1 · mM−1 depending on the experimental conditions used (Fig. 5)]. The variation in KI values, but not kinact values, is generally consistent with previous reports investigating the impact of dilution (Van et al., 2006; Parkinson et al., 2011). We propose that the quasi-irreversible or irreversible metabolism-dependent inhibition of CYP2C19 by omeprazole (rather than reversible inhibition) explains, at least in part, the observed clinical interactions between omeprazole (and esomeprazole) and CYP2C19 substrates, including clopidogrel, notwithstanding the considerable debate surrounding the role of CYP2C19 in the activation of the latter drug (Bouman et al., 2011; Sibbing et al., 2011).
Why Did Previous In Vitro Studies Miss the Metabolism-Dependent Inhibition of CYP2C19 by Omeprazole?
The IC50 or Ki values for inhibition of CYP2C19 in HLM by omeprazole reported in the literature range from 150 to 1 μM in nearly 40 studies (MTDI database). The lowest values (1 and 4 μM) were reported with some of the longest substrate incubation periods (60–120 min; presumably used because of the low turnover of S-mephenytoin), and few (if any) studies specifically included a preincubation with NADPH-fortified HLM to examine the possibility of metabolism-dependent inhibition of CYP2C19. On the basis of our data (Fig. 5), 60 to 120 min would provide ample time for inactivation of CYP2C19 by omeprazole during the substrate incubation, therefore leading to “unintentional” metabolism-dependent inhibition and artificially low IC50 or Ki values, as recently discussed (Parkinson et al., 2011). Our data also show that the IC50 shift diminishes as higher protein concentrations and longer substrate incubation times are used (Fig. 2b), probably because of the combination of inhibitor depletion (Fig. 2e) and the unintentional metabolism-dependent inhibition during the long substrate incubation (Parkinson et al., 2011).
Would We Predict Clinically Relevant CYP2C19 Inhibition?
As noted in the Introduction, we would not predict clinically significant inhibition of CYP2C19 by omeprazole on the basis of competitive inhibition alone. A thorough prediction of the effect of omeprazole on CYP2C19 substrates on the basis of the experimentally determined KI and kinact values necessitated the use of physiologically based pharmacokinetic modeling to allow for dynamic simulation of changes to omeprazole (including self-inhibition), substrate concentrations, and enzyme turnover (i.e., a so-called MDM), with the MSM (eq. 1) used as a comparator.
In the MDM under conditions in which in vitro inhibitor depletion and microsomal protein binding of omeprazole were minimal (Fig. 2, e and f; i.e., KI = 1.7 μM, Fig. 5d), the level of active CYP2C19 is predicted to decrease to ∼10% of baseline after approximately 7 days of omeprazole administration (Fig. 8a), which could cause up to a 10-fold increase in the AUC of compounds predominantly metabolized by CYP2C19. When the higher estimate of KI is used in the MDM (obtained with a 25-fold dilution under conditions in which significant inhibitor depletion occurs; Fig. 2e), the level of active CYP2C19 is predicted to decrease to ∼60% of baseline after approximately 12 days of omeprazole administration (Fig. 8b). Although S-mephenytoin is a model CYP2C19 probe substrate, only a 1.45- to 5.46-fold increase in its AUC (depending on KI) is predicted by the MDM after 14 days of omeprazole administration (40 mg twice daily), partly because of the contribution of other enzymes to its metabolism (e.g., CYP2B6) and possibly because of less-than-complete inactivation of CYP2C19, and/or possibly due to the inhibitory contribution of metabolites (e.g., omeprazole sulfone), not accounted for in the simulation.
In the MSM, when the lowest value of KI is used, omeprazole is predicted to cause a 2.85-fold increase in the AUC of a drug such as moclobemide, which has an fmCYP2C19 of 0.72 (Yu et al., 2001). The predicted AUC increase with moclobemide falls to 1.96-fold with the KI value of 9.1 μM. If the fmCYP2C19 were equal to 1 and the lowest KI value was used, then omeprazole is predicted to cause up to a 10.4-fold increase in the AUC of such a hypothetical “perfect” CYP2C19 substrate. However, because there is up to a 14.6-fold increase in the AUC of omeprazole in CYP2C19 PMs relative to EMs (Furuta et al., 1999), this prediction suggests that omeprazole does not completely inactivate CYP2C19, which is consistent with either scenario in the MDM after several days of administration of omeprazole (Fig. 8).
The clinical example of moclobemide (fmCYP2C19 = 0.72) is generally in agreement with these predictions, with AUC∞ increases averaging 2.21-fold (1.03- to 3.39-fold) in CYP2C19 EMs with omeprazole coadministration (Yu et al., 2001). The AUC increases predicted with the lowest KI in the MSM value fall within this observed range. In addition, to achieve the recently reported 1.39-fold increase in the AUC of clopidogrel with concomitant dosing of omeprazole (Angiolillo et al., 2011), the fmCYP2C19 for clopidogrel would need only to be approximately 0.31 according to the MSM.
Given the large difference in the predicted active CYP2C19 after several days of omeprazole administration in the MDM depending on which KI value is used (Fig. 8), the importance of determining the KI value under conditions at which microsomal protein binding and inhibitor depletion are minimized is underscored, similar to the case with IC50 values (Parkinson et al., 2011).
Is There Clinical Evidence of CYP2C19 Inhibition by Omeprazole?
In vivo evidence for the metabolism-dependent inhibition of CYP2C19 by omeprazole has already been reported. For instance, Klotz, 2006 reported that the healing rates of gastroesophageal reflux disease after 4 weeks of therapy with esomeprazole were not dependent on CYP2C19 status, as they are for lansoprazole. On the basis of the metabolite ratios of 5′-hydroxyomeprazole (formed by CYP2C19) to omeprazole sulfone (formed by CYP3A4), it was concluded that CYP3A4 plays the major role in the metabolism of esomeprazole after multiple dosing, consistent with autoinhibition of CYP2C19 and conversion of CYP2C19 EMs to intermediate metabolizers or PMs after repeat dosing (Klotz, 2006). The changes in metabolite ratio suggest that the effect of multiple dosing on omeprazole reflects the autoinhibition of CYP2C19, rather than increased stability due to higher gastric pH as originally suspected, especially because such time-dependent changes do not occur with other PPIs. On the other hand, the prescribing information for Nexium states “at repeated once-daily dosing with 40 mg, the systemic bioavailability is approximately 90% compared to 64% after a single dose of 40 mg” (Nexium prescribing information, 2010, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021153s036,021957s009,022101s006lbl.pdf). In addition, Andersson and Weidolf (2008) reported that when 15 mg of omeprazole (i.e., the racemate), esomeprazole, or R-omeprazole was administered orally for 7 days, exposure to esomeprazole (plasma AUC) increased by approximately 2-fold over 7 days, whereas exposure to omeprazole increased by only 52%, and exposure to R-omeprazole actually decreased by 9%. These results are consistent with our in vitro results showing that the inhibition of CYP2C19 by R-omeprazole appears to be largely reversible, whereas that of the racemate and esomeprazole are largely irreversible (Fig. 7).
In vivo evidence for metabolism-dependent inhibition of CYP2C19 by omeprazole also comes from clinical DDI studies with omeprazole as the perpetrator. For instance, in CYP2C19-extensive metabolizers (but not in poor metabolizers), the AUC of moclobemide increased by approximately 31% after a single 40-mg dose of omeprazole, but it increased by 121% after 8 days of dosing with 40 mg of omeprazole (Yu et al., 2001). Such an apparent increase in the exposure of a victim drug with repeated dosing of the perpetrator drug is often apparent with MDIs. In addition, omeprazole (but not lansoprazole or pantoprazole) has long been known to inhibit the metabolism of diazepam in vivo, and this inhibition occurs in CYP2C19 EMs but not PMs, further suggesting that the mechanism involves CY2C19 inhibition by omeprazole (Andersson et al., 1990; Lefebvre et al., 1992; Gugler et al., 1996).
Does Metabolism-Dependent Inhibition of CYP2C19 by Omeprazole Explain the PPI-Clopidogrel Interaction?
Our data and the predictions detailed above may explain, at least in part, the interaction between omeprazole (or esomeprazole) and clopidogrel. As noted in the Introduction, the FDA specifically warns against coadministration of clopidogrel and omeprazole (2010, http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm). Given that the in vivo half-life of omeprazole (and other PPIs) is short, and plasma protein binding is high, it is remarkable that many recent publications attribute the clopidogrel-omeprazole interaction to competitive (reversible) inhibition of CYP2C19 by omeprazole, with some suggestion that separation of dosing can prevent the interaction (Abraham et al., 2010; Tran et al., 2010; Bates et al., 2011). However, it should be noted that for clopidogrel, genetic differences in the metabolism or transport of the drug and in the therapeutic target (the P2Y12 receptor on platelets), as well as environmental factors (e.g., diet, disease, coadministered drugs), have been implicated in the variation in its clinical effect (for recent reviews see Abraham et al., 2010; Society for Cardiovascular Angiography and Interventions et al., 2010; Tran et al., 2010; Bates et al., 2011).
In addition, the interaction between omeprazole (or esomeprazole) and clopidogrel is particularly complex, as noted by Zhang et al. (2009). These authors note that clopidogrel itself is an MDI of CYP2C19, increasing the ratio of 5′-hydroxyomeprazole to omeprazole by ∼75% in CYP2C19 EMs (Chen et al., 2009; Zhang et al., 2009), and that clopidogrel and its 2-oxo metabolite (the precursor to the active metabolite) also directly inhibit CYP2C19 with IC50 values ≤1 μM. The authors suggested that the “stronger” effect of omeprazole on CYP2C19 may be due to the “time-dependent” inhibition we reported in our preliminary work (Paris et al., 2008; Zhang et al., 2009). Our finding of irreversible or quasi-irreversible inactivation of CYP2C19 by omeprazole, with up to a predicted 90% decrement in active CYP2C19 after approximately 7 days of dosing (Fig. 8a), is consistent with this hypothesis. The fact that the FDA warning applies only to omeprazole (http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm) and not the other PPIs is consistent with a lack of metabolism-dependent inhibition by the other PPIs examined in this study.
Given the minor inhibitory effects of omeprazole on other P450 enzymes (see supplemental data), the metabolism-dependent inhibition of CYP2C19 by omeprazole and the reports of clinical interactions are consistent with a significant role for CYP2C19 in the metabolism of clopidogrel. In addition, as described in the Introduction, direct inhibition of CYP2C19 by other PPIs is not likely to be the cause of a clinically significant interaction with clopidogrel, which is consistent with the lack of in vitro metabolism-dependent inhibition of CYP2C19 by lansoprazole and pantoprazole reported here and a lack of clinically significant pharmacokinetic interactions between either lansoprazole or pantoprazole and clopidogrel (MTDI database).
Potential Mechanisms of Inactivation of CYP2C19 by Omeprazole.
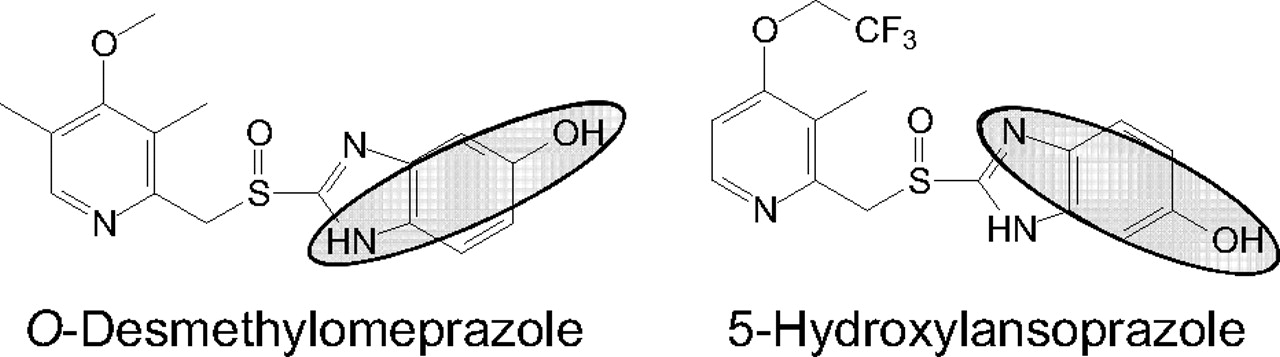
Follow-up studies will be needed to further elucidate the mechanism of inactivation of CYP2C19 by omeprazole (or esomeprazole). However, a few possibilities for the mechanism of CYP2C19 inactivation by omeprazole (or esomeprazole) are suggested by the reported metabolism of the individual enantiomers of omeprazole (Fig. 9, adapted from Abelö et al., 2000 and Andersson and Weidolf, 2008). Methylhydroxylation of omeprazole to form 5′-hydroxyomeprazole could involve the intermediacy of a benzylic radical and heme alkylation, analogous to the inactivation of CYP2C8 by gemfibrozil glucuronide (Ogilvie et al., 2006; Baer et al., 2009). The formation of 5-O-desmethylomeprazole (a para-aminophenol) could lead to a reactive quinoneimine that could inactivate CYP2C19 if formed in its active site. The latter possibility may be unlikely given that the 5-hydroxylation of lansoprazole also leads to para-aminophenol formation (Fig. 10; admittedly a tautomer of the analogous 5-O-desmethylomeprazole para-aminophenol), and yet lansoprazole is not an MDI of CYP2C19 (Fig. 4a).

Metabolic scheme for omeprazole enantiomers. The conversion of each enantiomer of omeprazole to the major metabolites and enzymes responsible for each is shown. The scheme is adapted from that published by Andersson and Weidolf, 2008, which is in turn based on in vitro data published by Abelö et al., 2000, which is the source of the CLint values.

Para-aminophenol formation from omeprazole and lansoprazole. The major metabolites of omeprazole (O-desmethylomeprazole) and lansoprazole (5-hydroxylansoprazole) are shown. The highlighted area indicates the part of the molecules that are potentially reactive para-aminophenols.
However, the data presented in Fig. 7 suggest that esomeprazole has a greater metabolism-dependent inhibition effect on CYP2C19 than does R-omeprazole. These data are in fact consistent with clinical observations in which the plasma Cmax of esomeprazole (40 mg daily of an oral solution) increases from 3.07 to 4.86 μM from day 1 to day 5, and that of omeprazole (40 mg daily of an oral solution) increases from 2.32 to 3.87 μM, whereas that of R-omeprazole (40 mg daily of an oral solution) only increases from 1.62 to 1.98 μM (Hassan-Alin et al., 2005). In addition, published in vitro results in HLM suggest that the CLint for esomeprazole sulfoxidation (catalyzed by CYP3A4) is 4.6-fold greater than that for R-omeprazole (Abelö et al., 2000), although the total CLint for R-omeprazole is approximately 3-fold higher than for esomeprazole. This finding is even more apparent in the clinical data, which show an approximately 14-fold higher AUC for the sulfone when esomeprazole is administered for 1 day (20 or 40 mg) than for R-omeprazole; this ratio increases to nearly 40-fold after 5 days of dosing (Hassan-Alin et al., 2005) because CYP3A4 plays a more important role in esomeprazole metabolism after multiple dosing (Klotz, 2006). Because we found that omeprazole sulfone is also an MDI of CYP2C19, it is possible that the combination of effects from esomeprazole and its sulfone explain the much greater inactivation of CYP2C19 by esomeprazole than R-omeprazole, and this possibility needs to be followed up.
At the same time, the CLint for the 5′-hydroxylation of R-omeprazole is approximately 10-fold higher than for esomeprazole, which, along with our results (Fig. 7), suggest that unless there is also a difference in the ultimate fate of R- versus S-5′-hydroxyomeprazole (e.g., partition ratio, benzylic radical formation, and oxygen rebound or other inactivating pathways), this pathway may not explain the formation of a reactive metabolite that inactivates CYP2C19. Enantiomer-enantiomer interactions at the active site of CYP2C19 when the racemate is used (as in some experiments in this study) could complicate the interpretation of results with the single enantiomers as previously described (Li et al., 2005), especially considering that the presence of R-omeprazole acts as an alternative substrate and offers substrate-protection for the irreversible inactivation by esomeprazole or its sulfone.
In conclusion, we have shown that omeprazole (but not pantoprazole or lansoprazole) is an MDI of CYP2C19 in HLM, cryopreserved human hepatocytes, and recombinant human CYP2C19. On the basis of the KI and kinact values for the metabolism-dependent inhibition of CYP2C19 by omeprazole in HLM, we predict that this inactivation is clinically significant. Furthermore, we provided evidence that esomeprazole is more likely to irreversibly inactivate CYP2C19 than is R-omeprazole. These findings have implications for the ongoing debate surrounding the interaction between clopidogrel (as well as other CYP2C19 substrates) and omeprazole and, in particular, esomeprazole.
Authorship Contributions
Participated in research design: Parkinson, Kazmi, Buckley, Paris, and Ogilvie.
Conducted experiments: Yerino, Ogilvie, and Kazmi.
Contributed new reagents or analytic tools: Toren and Rostami-Hodjegan.
Performed data analysis: Yerino, Ogilvie, Kazmi, Toren, Buckley, and Rostami-Hodjegan.
Wrote or contributed to the writing of the manuscript: Ogilvie, Parkinson, Kazmi, Yerino, Rostami-Hodjegan, Toren, and Buckley.
Acknowledgments
We thank the Hepatocellular Products and Services Department of XenoTech, LLC (Lenexa, KS) for providing historical data on CYP2C19 activity and the effect of 3-day omeprazole treatment on hepatocytes.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.041293.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- PPI
- proton pump inhibitor
- AUC
- area under the plasma concentration versus time curve
- CLint
- in vitro intrinsic clearance
- Cmax
- maximum plasma concentration
- P450
- cytochrome P450
- DDI
- drug-drug interaction
- EM
- extensive metabolizer
- HLM
- human liver microsomes
- Ki
- inhibition constant
- KI
- inhibitor concentration that supports half the maximal rate of inactivation
- kinact
- maximal rate of enzyme inactivation
- LC/MS-MS
- liquid chromatography/tandem mass spectrometry
- MTDI
- University of Washington Metabolism and Transport Drug Interaction database
- MDI
- metabolism-dependent inhibitor
- FDA
- U.S. Food and Drug Administration
- DMSO
- dimethyl sulfoxide
- PM
- poor metabolizer
- MSM
- mechanistic state model
- MDM
- mechanistic dynamic model
- amu
- atomic mass units.

- Received June 22, 2011.
- Accepted July 27, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}