


Abstract
During drug development, it is important to predict the activities of multiple metabolic enzymes, not only cytochrome P450 (P450) but also non-P450 enzymes, such as conjugative enzymes and aldehyde oxidase (AO). In this study, we focused on prediction of AO-mediated human metabolism and pharmacokinetics (PK) of 6-(2-amino-4-phenylpyrimidine-5-yl)-2-isopropylpyridazin-3(2H)-one (FK3453) (Astellas Pharma Inc.), the development of which was suspended due to extremely low exposure in human, despite good oral bioavailability in rat and dog. We examined species difference in oxidative metabolism of the aminopyrimidine moiety of FK3453, catalyzed by AO, using human-chimeric mice with humanized liver (h-PXB mice) and rat-chimeric mice (r-PXB mice) transplanted with rat hepatocytes. AO activity of h-PXB mouse hepatocytes was higher than that of r-PXB mouse hepatocytes. Moreover, higher concentrations of human-specific AO-generated FK3453 metabolite A-M were detected in urine and feces after administration of FK3453 to h-PXB mice versus r-PXB mice. The total clearance of h-PXB mice was 2-fold higher than that of r-PXB mice. These results agreed reasonably well with the metabolism and PK profiles of FK3453 in human and rat. Our results indicated that h-PXB mice should be helpful for predicting the metabolic profile of drugs in humans, and the use of both h-PXB and r-PXB mice should be helpful for evaluation of species differences of AO metabolic activity.
Introduction
It is important to predict drug metabolism and pharmacokinetics (PK) in human during the preclinical stage of pharmaceutical development, because new drug candidates with diverse chemical structures may be metabolized by not only cytochrome P450 (P450), but also non-P450 enzymes, such as UDP-glucuronosyltransferase (UGT), sulfotransferase, aldehyde oxidase (AO), and xanthine oxidase (Beedham, 1997). In recent years, the drop-out rate during drug development has been decreasing as a result of improved predictability of human drug metabolism and PK parameters (Kola and Landis, 2004). However, it is still difficult to predict non-P450 metabolism, especially involving AO.
AO is a molybdoflavoprotein (Beedham, 1987; Garattini et al., 2003) that catalyzes the metabolism of not only aldehydes but also nitrogenous heterocycles; it is involved in the oxidative metabolism of drugs such as 6-deoxypenciclovir, 6-mercaptopurine, 5-fluoro-2-pyrimidinone, methotrexate, and zaleplon (Guo et al., 1995; Rashidi et al., 1997; Kawashima et al., 1999; Kitamura et al., 1999; Lake et al., 2002; Rashidi et al., 2007). Moreover, in the presence of its electron donor, AO can mediate the reduction of a variety of compounds, such as sulfoxides, N-oxides, and nitrosamines (Kitamura et al., 2006). AO activity shows significant species differences due to the presence of multiple genes coding for AO isoforms (Garattini and Terao, 2011).
6-(2-Amino-4-phenylpyrimidine-5-yl)-2-isopropylpyridazin-3(2H)-one (FK3453) is a drug developed as a novel adenosine A1/2 dual inhibitor for the treatment of Parkinson's disease by Astellas Pharma Inc., Ibaraki, Japan (Akabane et al., 2011), but its development was suspended due to extremely low systemic exposure in a clinical study, despite encouraging results in animal experiments. FK3453 is metabolized by AO, and the oxidative metabolite of the aminopyrimidine moiety was identified as the major metabolite in plasma after dosing of FK3453 in human. Akabane et al. (2011) suggested that the formation of the AO-generated metabolite (A-M), [6-(2-amino-6-oxo-4-phenyl-1,6-dihydropyrimidin-5-yl)-2-isopropyl pyridazin-3(2H)-one], accounted for the poor oral bioavailability in humans. Species differences of metabolism and disposition resulted in the failure to predict the poor results in human.
Chimeric mice with humanized liver, generated using urokinase-type plasminogen activator (uPA+/+)/severe combined immunodeficiency (SCID) mice repopulated with human hepatocytes (h-PXB mice; Phonixbio, Co., Ltd., Hiroshima, Japan) have been reported (Tateno et al., 2004). The expression levels and metabolic activities of P450 and non-P450 enzymes in liver of h-PXB mice were similar to those in human (Katoh et al., 2004, 2005), and human-specific metabolites were detected in h-PXB mice (Inoue et al., 2009; Kamimura et al., 2010; Yamazaki et al., 2010; Serres et al., 2011). Thus, h-PXB mice could be a good in vivo model for predicting drug metabolism in humans. In addition to h-PXB mice, rat-chimeric mice (r-PXB mice) containing high levels of rat hepatocytes have been used to compare metabolism between rats and humans (Tateno et al., 2004; Emoto et al., 2005). Yamazaki et al. (2010) reported that human-specific metabolites of the pyrazolopyrimidine moiety could be distinguished from rat metabolites by using h-PXB mice and r-PXB mice.
In this study, we performed both in vitro and in vivo investigations using h-PXB mice and r-PXB mice to evaluate the usefulness of these models for predicting human metabolism and PK of FK3453.
Materials and Methods
Chemicals.
FK3453 and its metabolite, A-M, were supplied by Astellas Pharma Inc. All other reagents and solvents were commercial products of the highest available grade or analytical grade.
Animals.
h-PXB mice and r-PXB mice, in which the endogenous hepatocytes had been replaced with human hepatocytes and rat hepatocytes, respectively, were prepared by PhoenixBio Co. Ltd. according to the method described previously (Tateno et al., 2004; Emoto et al., 2005). Hepatocytes of a human donor (African American male, 5 years old) for transplantation to prepare h-PXB mice were obtained from BD Biosciences (San Jose, CA). Rat hepatocytes for transplantation to prepare r-PXB mice were isolated from liver of Sprague-Dawley (SD) rats (4 weeks of age, male).
The extent of replacement of host hepatocytes with human hepatocytes or rat hepatocytes, calculated as replacement index (RI), was determined by measurement of the concentration of human or rat albumin in blood collected from the tail vein of each PXB mouse. The RI was estimated by the correlation curve between the human albumin levels in mouse blood and determined by using human-specific cytokeratin 8/18-immunostained liver sections (Tateno et al., 2004). Average RI values of h-PXB mice and r-PXB mice used in this study were 83% and nearly 100%, respectively, as estimated from the corresponding albumin level in blood.
The h-PXB mice and r-PXB mice were housed in a temperature- and humidity-controlled environment under a 12-h light/dark cycle with free access to tap water and diet (h-PXB mice, CRF1 diets containing vitamin C; r-PXB mice, normal diet). All animal studies were approved by the institutional animal ethics committee and conducted in accordance with the regulations on the use of living modified organisms at each facility.
Isolation and Purification of Hepatocytes from h-PXB Mice and r-PXB Mice.
Hepatocytes were isolated from h-PXB mice and r-PXB mice (13 and 12 weeks of age, respectively) using in situ collagenase perfusion (Yamasaki et al., 2010). h-PXB mice hepatocytes (h-hepatocytes) originally contained approximately 13% of mouse hepatocytes, although r-PXB mice hepatocytes (r-hepatocytes) were almost free of mouse hepatocytes. Therefore, we used h-hepatocytes purified from chimeric hepatocytes of h-PXB mice by the use of 66Z rat IgG and magnetic beads bearing anti-rat IgG antibodies. The magnetic removal of mouse hepatocytes reduced the level of mouse hepatocytes to approximately 2%; in this study, purity of human hepatocytes values of h-hepatocytes ranged from 97.4 to 98.3%. Cell viability of hepatocytes used in experiments was more than 78 to 91% (trypan blue exclusion test).
In Vitro Metabolic Study Using h-Hepatocytes and r-Hepatocytes.
Hepatocyte suspension (1 × 106 cells /ml) was incubated in Krebs-Henseleit buffer without serum after treatment with 10 μM FK3453 at 37°C under an atmosphere of 5% CO2/95% O2. The final concentration of acetonitrile was 0.5% (v/v) in the reaction mixture. The plates (24 wells) were shaken gently with an orbital shaker. The incubation mixtures were collected at 0, 0.25, 0.5, 1, and 2 h after treatment and frozen in liquid nitrogen. Samples were thawed for analysis and spiked with two volumes of acetonitrile. After centrifugation, aliquots of the supernatant were subjected to liquid chromatography/tandem mass spectrometry (LC/MS/MS).
Administration of FK3453.
FK3453 solution was administered to PXB mice at a single dose of 3 mg/kg. FK3453 was formulated in 10% dimethyl sulfoxide, 10% PEG400 with an equivalent amount of hydrochloride in saline for intravenous dosing, and in 50% PEG400 with an equivalent amount of hydrochloride in saline for oral dosing. Blood samples were collected from the orbital vein of PXB mice during 0 to 6 h postdose using heparinized glass. Plasma samples separated after centrifugation were stored at −30°C.
Urinary and Fecal Excretion.
Pooled urine and feces after administration were collected during the period of 0 to 24 h. Each metabolic cage was washed with water after collection of urine and feces. Samples of urine and feces were homogenized with 10 volumes of water, and the homogenates were stored at −30°C.
Plasma Protein Binding.
Plasma protein binding ratio of FK3453 (1 μg/ml) in h-PXB and r-PXB mice was determined by equilibrium dialysis after incubation of each plasma with FK3453.
Determination of Metabolite Profile.
Hepatocyte suspension after 2-h incubation (150 μl), urine (150 μl), 10% of fecal homogenate (200 μl), and pooled plasma (50 μl) were mixed with equivalent volumes of acetonitrile and centrifuged at 14,000g for 5 min. The supernatants were subjected to LC/MS/MS.
Aliquots of 5 μl were introduced into a Acquity UPLC system (Waters, Milford, MA) with a Waters Acquity BEH C18 column, 1.7 μm, 2.1 × 100 mm. The mobile phase consisted of 5 mM ammonium formic acid containing 5% acetonitrile (solvent A) and acetonitrile (solvent B). The flow rate was 0.2 ml/min. The starting condition for the gradient was 100:0 (A/B) until 2.5 min, then the mobile phase composition was changed linearly to 65:35 (A/B) from 2.5 min to 25 min, and 10:90 (A/B) was maintained until 27 min. The gradient was returned to 100:0 (A/B) from 27 min to 27.01 min for re-equilibration. UV detection at 254 nm was used. The retention times of FK3453 and A-M were 19.9 min and 16.3 min, respectively. The MS/MS experiments were performed on a Thermo LTQ Orbitrap Velos (Thermo Fisher Scientific, Waltham, MA).
The parameters for the ESI source were as follows: capillary temperature 330°C; spray voltage 4.5 kV (positive ion mode) or 4.0 kV (negative ion mode); sheath gas flow rate 40 arbitrary unit (nitrogen gas); and auxiliary gas flow rate 10 (nitrogen gas). The mass spectrometer was operated in both positive and negative ion modes. The ions were monitored from m/z 150 to 800. Normalized collision energies for MS2, MS3, and MS4 were set at 35, 40, and 30%, respectively.
Quantitation of FK and Its Metabolite, A-M.
Hepatocyte suspension after 2-h incubation (20 μl), urine (20 μl), and plasma (15 μl) were each mixed with two volumes of acetonitrile and internal standard solution (carbamazepine). The supernatants after centrifugation at 14,000g for 5 min were injected into the LC/MS/MS. Ten percent of fecal homogenates (200 μl) were extracted with 5 ml of t-butylmethylether and internal standard solution (carbamazepine). The organic layer (4 ml) was evaporated to dryness, and the residues were dissolved in aqueous acetonitrile (200 μl). Aliquots of 10 μl were applied to an Inertsil ODS-3 (3 μm, 50 × 2.1 mm) column on an HP-1100 series HPLC instrument (Agilent Technologies, Santa Clara, CA) at 40°C. The mobile phase was composed of 10 mM ammonium acetate (solvent A) and acetonitrile (solvent B), and the flow rate was 0.2 ml/min. The isocratic condition for HPLC was 70:30 (A/B). The MS/MS experiments were conducted by using an API2000 LC/MS/MS system (Applied Biosystems, Foster City, CA). Mass numbers of molecular and product ions used for identification of FK3453 and A-M were as follows: FK3453 m/z = 308.1 [M+H]+ to 265.9, A-M m/z = 324.1 [M+H]+ to 281.8. The retention times of FK and A-M were 3.6 and 1.6 min, respectively.
Determination of PK Parameters.
Pharmacokinetic parameters were determined by noncompartmental methods using the concentration-time curve profile. The maximum plasma concentration (Cmax) and the time at which the maximum concentration were achieved (tmax), which were determined from actual values. The area under the plasma concentration-time curve (AUC) was calculated from the time course using trapezoidal extrapolation from the last quantifiable time to infinity. The total clearance (CLt) after intravenous administration (i.v.) and the oral clearance (CLoral) after oral administration (p.o.) were calculated as Dose/AUCiv and Dose/AUCpo, respectively. The terminal elimination half-life (t1/2) was estimated as ln2/slope, where the slope is that of the plot of the terminal elimination phase on a logarithmic scale. The oral bioavailability (F) was calculated as AUCpo/AUCiv.
Calculation of In Vitro Intrinsic Clearance.
In vitro intrinsic clearances (CLint, in vitro) were calculated from the time course of disappearance of unchanged compound after incubation with hepatocytes of h-PXB mice and r-PXB mice. The disappearance rate constant of unchanged drug was calculated from each plot fitted to the first-order elimination rate constant for FK3453.
Results
In Vitro Metabolic Study Using Fresh Hepatocytes Isolated from h-PXB and r-PXB Mice.
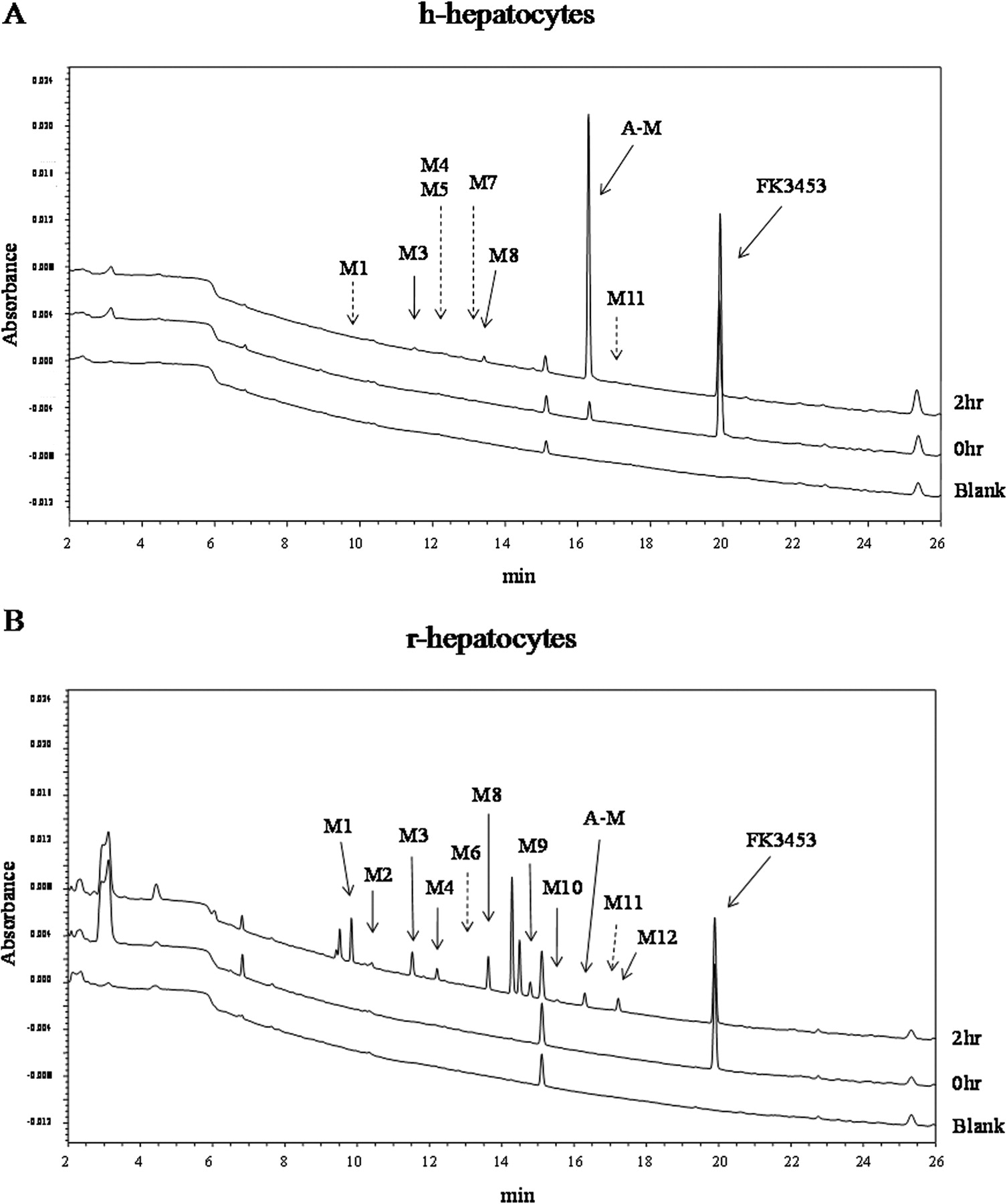
Chemical structures of FK3453 and its AO-generated metabolite, A-M, are shown in Fig. 1. AO was confirmed to be responsible for A-M formation by means of an in vitro experiment using AO inhibitor with liver cytosol (Akabane et al., 2011). Figure 2 shows depletion profiles of FK3453 up to 2 h during incubation with fresh hepatocytes isolated from PXB mice. Loss of FK3453 was observed during incubation with both h-hepatocytes and r-hepatocytes. CLint, in vitro in h-hepatocytes and r-hepatocytes were 10 and 5 μl · min−1 · 106 cells−1, respectively, calculated from the first-order elimination rate constants of FK3453 on a logarithmic scale. Formation of A-M increased, concomitantly with the disappearance of FK3453. The formation of A-M after 2-h incubation with h-hepatocytes was 15-fold higher than in the case of r-hepatocytes (Table 1). Although A-M was the predominant metabolite with human hepatocytes, not only A-M but also several other peaks were detected with both h-hepatocytes and r-hepatocytes. However, few metabolites were detected with h-hepatocytes, whereas many metabolites besides A-M were found with r-hepatocytes (Fig. 3; Table 1).

Chemical structures of FK3453 and its AO metabolite (A-M).

Time courses of FK3453 (closed square) and A-M (closed triangle) during 2-h incubation with h-hepatocytes and r-hepatocytes isolated from h-PXB and r-PXB mice, respectively. FK3453 (10 μM) was incubated for 2 h with h-hepatocytes (left) and r-hepatocytes (right) at 1 × 106 cells/ml. Each point and bar represents the mean ± S.D. (n = 3).
In vitro and in vivo (i.v./p.o.) comparison of metabolic profiles of FK3453 in h-PXB mice and r-PXB mice

UV chromatograms of metabolites after incubation of FK3453 with h-hepatocytes (A) and r-hepatocytes (B). Pooled samples (n = 3) of blank, 0 and 2 h after incubation with h-hepatocytes were chromatographed with detection at 254 nm. Solid arrows indicate metabolites detected by UV and mass spectrometry. Dotted arrows show metabolites detected by only mass spectrometry.
Metabolites of FK3453 in Urine, Feces, and Plasma of h-PXB and r-PXB Mice.
Plasma, urine, and feces pooled during 24 h after i.v./p.o. administration of FK3453 were collected. Excretions of FK3453 in urine and feces amounted to the range from 0.1 to 1.1% of the dose in h-PXB mice and r-PXB mice, which were very low. The metabolites found in plasma were similar to those of urine, and feces in both h-PXB mice and r-PXB mice, except for M11. In contrast, A-M was the predominant metabolite in urine and feces of h-PXB mice, whereas urinary and fecal excretion of A-M was less in r-PXB mice. Other metabolites besides A-M were found in both h-PXB mice and r-PXB mice. In particular, M6, M9, M10, and M12 were specifically detected in plasma, urine, and feces of r-PXB mice but not h-PXB mice (Fig. 4; Table 1).

UV chromatograms of FK3453 metabolites from urine (A and C) and feces (B and D) of h-PXB mice (A and B) and r-PXB mice (C and D). Pooled 24-h samples (n = 3) of blank, and after i.v., and p.o. administration to h-PXB mice and r-PXB mice (n = 3) were chromatographed with detection at 254 nm. Solid arrows indicate metabolites detected by UV and mass spectrometry. Dotted arrows show metabolites detected by only mass spectrometry.
PK Profile of FK3453 in h-PXB Mice and r-PXB Mice.
Plasma concentrations and PK parameters after i.v./p.o. administration of FK3453 at 3 mg/kg to h-PXB mice and r-PXB mice are shown in Fig. 5 and Table 2. The values of CLt after i.v. administration in h-PXB mice and r-PXB mice were 70.7 and 34.0 ml · min−1 · kg−1, respectively. On other hand, the values of CLoral after p.o. administration were 234.2 and 61.6 ml · min−1 · kg−1, respectively. Elimination was faster and systemic exposure to FK3453 was lower in h-PXB mice, compared with r-PXB mice. Furthermore, bioavailability of FK3453 after oral dosing in h-PXB mice was lower than that in r-PXB mice.

Plasma concentration-time curves of FK3453 in h-PXB mice and r-PXB mice. Data are mean ± S.D. (n = 3∼4).
Comparison of PK parameters of FK3453 among PXB mice, human, and rat
The value of CLoral calculated from phase I clinical trial data at 10 mg p.o. in human was even higher than that in h-PXB mice. In contrast, the values of CLoral measured in r-PXB mice were similar to those in rats dosed at 3.2 mg/kg (Akabane et al., 2011).
Plasma Protein Binding of FK3453.
Plasma binding ratios of FK3453 in h-PXB mice and r-PXB mice were 79.5 and 76.7%, respectively (Table 3). These values were similar to those evaluated in human and rat. The level of human albumin in plasma was 9.9 mg/ml in h-PXB mice, and that of rat albumin was 17.1 mg/ml in r-PXB mice (data not shown).
Summary of plasma protein binding in PXB mice, human, and rat
Data are mean ± S.D. of n = 3.
Discussion
Prediction of the metabolism and PK of new chemical entities in human is important in the early preclinical stage of pharmaceutical development. The number of new chemical entities metabolized by AO also seems to be increasing (Pryde et al., 2010), and this is important because AO activity shows species differences (Grattini et al., 2011). For example, zaleplon is metabolized by AO to 5-oxo-zaleplon, the major circulating metabolite in human, whereas deacetyl-zaleplon, a CYP3A metabolite, is the major metabolite in rats, mice, and dogs (Kawashima et al., 1999). Furthermore, human-specific AO metabolites of 6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4] triazolo[4,3-b]pyridazin-3-ylthio)quinoline (SGX523) were reported to cause acute renal failure in human (Diamond et al., 2010). Zhang et al. (2011) reported that the clinical development of 6-(2,4-difluoro-phenoxy)-2-((R)-2-hydroxy-1-methyl-ethylamino)-8-((S)-2-hydroxy-propyl)-8H-pyrido[2,3-d]pyrimidin-7-one (RO1), containing a pyridopyrimidine moiety, as a p38 kinase inhibitor candidate was terminated because of its rapid clearance by AO in human. In addition, famciclovir, methotrexate, zebularine, zoniporide, and other similar compounds are AO substrates (Rashidi et al., 1997; Kitamura et al., 1999; Klecker et al., 2006; Dalvie et al., 2010). In general, AO activity seems to be high in monkey and human, but low in rat and absent in dog. Zientek et al. (2010) established a method for clearance prediction using human liver cytosol and S9 fraction and obtained a good in vitro-in vivo correlation of CLint for compounds metabolized by AO. However, in vitro CLint was lower than in vivo CLint, probably owing to partial inactivation of AO during experimental procedures.
Whereas favorable PK profiles of FK3453 were observed in rats and dogs, systemic exposure of FK3453 in humans turned out to be very low. This finding was considered to be caused by high AO activity in human, and indeed, a high plasma concentration of A-M, the AO-catalyzed metabolite of FK3453, was detected in human (Akabane et al., 2011). A-M does not show pharmacological activity (data not shown).
In this study, we further examined species differences of AO activity toward FK3453 using h-PXB mice and r-PXB mice. The expression and activity of AO in liver of h-PXB mice have been reported (Kitamura et al., 2008). In contrast, the AO activity of r-PXB mice seems to be low, probably because rat hepatocytes for r-PXB mice were derived from Crj:SD rats, which show lower AO activity than other strains of rats (Moriyasu et al., 2006; Sugihara et al., 2006; Itoh et al., 2007). The levels of expression P450 and non-P450 enzymes and metabolic activities in fresh h-hepatocytes are similar to those of actual human hepatocytes (Yoshitsugu et al., 2006; Yamasaki et al., 2010).
CLint, in vitro calculated from the disappearance rate of FK3453 in h-hepatocytes was 2-fold greater than that in r-hepatocytes (Fig. 2), in agreement with the difference of CLt between h-PXB mice and r-PXB mice. Moreover, A-M was the predominant metabolite when FK3453 was incubated with h-hepatocytes. In contrast, other metabolites were generated instead of lower A-M formation in r-PXB mice (Figs. 2 and 3). We confirmed that A-M formation was mediated by AO in h-hepatocytes because this activity was inhibited by an AO inhibitor, menadione, but not by a P450 inhibitor, 1-aminobenzotriazole (data not shown). This is in agreement with the findings of Akabane et al. (2011).
In vivo, unchanged excretion of FK3453 was low in h-PXB mice and r-PXB mice. The metabolites found in plasma were approximately similar to those of urine, and feces in both h-PXB mice and r-PXB mice. A-M was also the major metabolite in urine and feces after i.v./p.o administration of FK3453 to h-PXB mice. In contrast, many metabolites were detected in plasma, urine, and feces of r-PXB mice. In particular, M6, M9, M10, and M12 were specifically detected in r-PXB mice but not in h-PXB mice. These metabolites would be formed by P450, considering each mass.
Furthermore, each in vitro metabolic profile was in agreement with in vivo metabolic profiles in h-PXB mice and r-PXB mice, respectively, although M2 observed in urine of h-PXB mice was not detected in h-hepatocytes. In this case, the levels of remaining mouse hepatocytes in liver of h-PXB mice might not influence in vitro and in vivo metabolic profiles except for M2, because h-hepatocytes almost consisted of human donor hepatocytes.
In the PK study, CLt or CLoral of h-PXB mice was 2-fold higher than that of r-PXB mice, in agreement with the CLint, in vitro values. High levels of A-M were observed in urine and feces of h-PXB mice after administration of FK3453 (Fig. 5; Table 1). The CLoral of FK3453 in h-PXB mice was not in agreement with that of human from a phase I clinical trial (Table 2). In contrast, both CLt and CLoral of FK3453 in r-PXB mice were similar to those in actual rats. This discrepancy may be due to the significant difference of hepatic blood flow rate between h-PXB mice and human, compared with the relationship between r-PXB mice and rats (Davies and Morris, 1993).
Plasma binding ratios of FK3453 in h-PXB mice and r-PXB mice agreed well with those in actual human and rat determined by equilibrium dialysis (Table 3). This result supports that high levels of human and rat albumins are expressed in blood of the PXB mice (Tateno et al., 2004). Therefore, the difference in A-M formation may be mainly due to species difference in AO, but not species difference of plasma binding.
In this study, we conducted PK study using h-PXB mice that contain approximately 20% of mice hepatocytes. It is also important to consider the contribution of the metabolism in remaining mice hepatocytes in hepatic of PXB mice. We examined metabolic activity of FK3453 using hepatocytes isolated from SCID mice compared with h-PXB mice. CLint, in vitro in SCID mouse hepatocytes and h-hepatocytes were 3 and 10 μl · min−1 · 106 cells−1, respectively. In both cases, the total amount of remaining FK3453 and formation of A-M after 2-h incubation with FK3453 is approximately 100%. These results suggested that AO activity of h-hepatocytes was 3-fold higher than that of SCID mouse hepatocytes (data not shown). Remaining m-hepatocytes in h-PXB mice would not affect human PK and metabolism. As a result, it is at least possible to predict from these data that CL of humans is higher than that of rats.
Furthermore, it is known of species differences of AO family that a single AO gene (AOX1) is coding in humans, four genes (Aox1, Aox3, Aox4, and Aox3l1) in mice genomes. AOX4 is highly presented in the Harderian gland and skin of mice, is absent in humans (Garattini et al., 2009; Terao et al., 2009). Evaluation of extrahepatic metabolism, which shows species differences, may increasingly improve the predictability of AO metabolism in human.
In conclusion, although h-PXB mice have some limitations for prediction of human drug metabolism, our results suggest that the combined use of h-PXB mice and r-PXB mice may be helpful for examining species differences of drug metabolism and for predicting human metabolism during the early stages of drug development.
Authorship Contributions
Participated in research design: Sanoh, Murai, Terashita, Teramura, and Ohta.
Conducted experiments: Sanoh and Nozaki.
Contributed new reagents or analytic tools: Murai, Terashita, and Teramura.
Performed data analysis: Sanoh.
Wrote or contributed to the writing of the manuscript: Sanoh, Nozaki, and Ohta.
Acknowledgments
We thank members in PhoenixBio Co. Ltd. for isolation of hepatocytes from PXB mice.
Footnotes
This work was supported by a Grant-in-Aid for Young Scientists (B) from Japan Society for the Promotion of Science [Grant 22790109].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
ABBREVIATIONS:
- PK
- pharmacokinetics
- UGT
- UDP-glucuronosyltransferase
- P450
- cytochrome P450
- AO
- aldehyde oxidase
- FK3453
- 6-(2-amino-4-phenylpyrimidine-5-yl)-2-isopropylpyridazin-3(2H)-one
- A-M
- AO-generated metabolite
- SCID
- severe combined immunodeficiency
- h-PXB mice
- chimeric mice with humanized liver
- r-PXB mice
- chimeric mice transplanted with rat hepatocytes
- SD
- Sprague-Dawley
- RI
- replacement index
- h-hepatocytes
- h-PXB mice hepatocytes
- r-hepatocytes
- r-PXB mice hepatocytes
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- PEG400
- polyethylene glycol 400
- HPLC
- high-performance liquid chromatography
- Cmax
- the maximum plasma concentration
- tmax
- the time at which Cmax was achieved
- AUC
- the area under the plasma concentration-time curve
- CLt
- total clearance
- i.v.
- intravenous administration
- CLoral
- oral clearance
- p.o.
- oral administration
- t1/2
- half-life
- F
- the oral bioavailability
- CLint, in vitro
- in vitro intrinsic clearance
- SGX523
- 6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinoline
- RO1
- 6-(2,4-difluoro-phenoxy)-2-((R)-2-hydroxy-1-methyl-ethylamino)-8-((S)-2-hydroxy-propyl)-8H-pyrido[2,3-d]pyrimidin-7-one.
- Received August 5, 2011.
- Accepted October 7, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}