


Abstract
Elucidation of the rate-determining process in the overall hepatic elimination of drugs is critical for predicting their intrinsic hepatic clearance and the impact of variation of sequestration clearance on their systemic concentration. The present study investigated the rate-determining process in the overall hepatic elimination of the HMG-CoA reductase inhibitors pravastatin, pitavastatin, atorvastatin, and fluvastatin both in rats and humans. The uptake of these statins was saturable in both rat and human hepatocytes. Intrinsic hepatic clearance obtained by in vivo pharmacokinetic analysis in rats was close to the uptake clearance determined by the multiple indicator dilution method but much greater than the intrinsic metabolic clearance extrapolated from an in vitro model using liver microsomes. In vivo uptake clearance of the statins in humans (pravastatin, 1.44; pitavastatin, 30.6; atorvastatin, 12.7; and fluvastatin, 62.9 ml/min/g liver), which was obtained by multiplying in vitro uptake clearance determined in cryopreserved human hepatocytes by rat scaling factors, was within the range of overall in vivo intrinsic hepatic clearance (pravastatin, 0.84-1.2; pitavastatin, 14-35; atorvastatin, 11-19; and fluvastatin, 123-185 ml/min/g liver), whereas the intrinsic metabolic clearance of atorvastatin and fluvastatin was considerably low compared with their intrinsic hepatic clearance. Their uptake is the rate-determining process in the overall hepatic elimination of the statins in rats, and this activity likely holds true for humans. In vitro-in vivo extrapolation of the uptake clearance using a cryopreserved human hepatocytes model and rat scaling factors will be effective for predicting in vivo intrinsic hepatic clearance involving active uptake.
Predicting the pharmacokinetic properties of drug candidates in preclinical stages of development has been a critical issue for avoiding failure in clinical stages of development because of pharmacokinetics. The liver is the major clearance organ for drugs in the body where the inactivation mechanisms are composed of metabolic enzymes and drug transporters. These inactivation mechanisms are associated with the hepatic first-pass effect after oral administration and with elimination from the systemic circulation. It is well accepted that, because of large species differences in drug metabolism, the results of animal studies cannot be directly extrapolated to humans. Instead, in vitro systems have been developed to replace animal studies and provide reliable predictions. In particular, human liver microsomes enable the reliable prediction of the metabolic clearance of drugs in the liver of humans (Iwatsubo et al., 1997; Obach, 1999; Naritomi et al., 2001; Stringer et al., 2008; Kilford et al., 2009). It has been found that the substrates of hepatic uptake transporters, organic anion-transporting polypeptide (OATP) 1B1 and OATP1B3, include anionic drugs whose major elimination pathway is metabolism by cytochrome P450 (P450) and UDP-glucuronosyltransferase in the liver. These drugs include cerivastatin, atorvastatin, fluvastatin, repaglinide, and telmisartan (Fischer et al., 1999; Jacobsen et al., 2000; Bidstrup et al., 2003; Kirchheiner et al., 2003; Shitara and Sugiyama, 2006). These drugs have been considered as outliers in the prediction of elimination using human liver microsomes because of active transport in their uptake process, concentrating substrate drugs inside the cells.
For these drugs, the impact of variation of sequestration clearance on the drug concentrations in the systemic circulation depends on a rate-determining process (Kusuhara and Sugiyama, 2009). Despite its importance for predicting the impact of variation of metabolic activity or canalicular efflux on systemic exposure, there are only a few studies examining the rate-determining process for the hepatic elimination of pravastatin and methotrexate in rats (Yamazaki et al., 1996; Ueda et al., 2001). Because of the lack of information regarding liver concentrations of these drugs, the rate-determining process in their overall hepatic elimination has not been investigated in humans. We proposed an in vitro-in vivo extrapolation in which the uptake clearance is extrapolated from an in vitro model using cryopreserved human hepatocytes and rat scaling factor based on the finding that scaling factors for P450-mediated metabolism are preserved across the species (Naritomi et al., 2001). The extrapolated uptake clearance of pravastatin was within the range of clinically reported intrinsic hepatic clearance, suggesting that the uptake is also the rate-determining process in humans (Watanabe et al., 2009). The purpose of this study was to apply this method to the other HMG-CoA reductase inhibitors (statins), pitavastatin, atorvastatin, and fluvastatin, in rats and humans. Hepatic elimination is the major pathway for elimination of these statins from the systemic circulation, but the mechanisms involved are different: atorvastatin and fluvastatin are metabolized by CYP3A4 and CYP2C9, respectively, and pitavastatin undergoes biliary excretion by breast cancer resistance protein (BCRP) (Fischer et al., 1999; Jacobsen et al., 2000; Kirchheiner et al., 2003; Hirano et al., 2005). Hepatic uptake of pravastatin, pitavastatin, and atorvastatin involves a transporter, OATP1B1, based on a kinetic analysis of pitavastatin using human hepatocytes (Hirano et al., 2004) and clinical studies for pravastatin, pitavastatin, and atorvastatin. Generic variation of OATP1B1, such as OATP1B1*5 and OATP1B1*15, shows reduced transport activities compared with the reference OATP1B1 (OATP1B1*1a) (Tirona et al., 2001; Iwai et al., 2004; Nozawa et al., 2005), and healthy volunteers carrying those genotypes exhibit greater systemic exposure of pravastatin, pitavastatin, and atorvastatin, indicating the importance of OATP1B1 in their hepatic uptake process (Nishizato et al., 2003; Maeda et al., 2006; Niemi et al., 2006; Ieiri et al., 2007; He et al., 2009). On the other hand, the systemic exposure of fluvastatin was independent of OATP1B1 genotype (Niemi et al., 2006), whereas fluvastatin is a substrate of OATP1B1 (Kopplow et al., 2005; Noé et al., 2007). OATP1B1 is suggested to only make a negligible contribution to the hepatic elimination of fluvastatin.
In the present study, the overall intrinsic hepatic clearances of the statins were determined from in vivo studies using rats, and their uptake clearances were determined using a multiple indicator dilution (MID) technique. Metabolic clearances were determined using liver microsomes. In addition, in vitro parameters for hepatic uptake and metabolism were determined using cryopreserved human hepatocytes and liver microsomes, and extrapolated to the corresponding in vivo parameters to compare these parameters with clinically determined intrinsic hepatic clearances. The present study suggests that hepatic uptake is the predominant factor for hepatic elimination of these representative statins.
Materials and Methods
Materials.
Pravastatin and a pravastatin analog, R-122798, were donated by Daiichi Sankyo Co. (Tokyo, Japan). Pitavastatin was donated by Kowa Co. (Tokyo, Japan). Atorvastatin was purchased from AK Scientific (Mountain View, CA). Fluvastatin and cerivastatin were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Fluorescein isothiocyanate dextran 4000 [(FD-4) 4000 Da] was purchased from Sigma-Aldrich (St. Louis, MO). All the other chemicals and reagents were of analytical grade and were readily available from commercial sources.
Animals.
Male Sprague-Dawley (SD) rats (6–7 weeks old) were purchased from Nippon SLC (Shizuoka, Japan). All the animals were maintained under standard conditions with a reversed light/dark cycle and were treated humanely. Food and water were available ad libitum. The studies were conducted in accordance with the guidelines of the Institutional Animal Care Committee, Graduate School of Pharmaceutical Sciences, The University of Tokyo (Tokyo, Japan).
Preparation of Rat and Human Hepatocytes.
Isolated rat hepatocytes were prepared from SD rats by the collagenase perfusion method described previously (Yamazaki et al., 1993). Isolated hepatocytes (viability >88%) were suspended in Krebs-Henseleit buffer, adjusted to 2.0 × 106 cells/ml, and stored on ice before the uptake experiment. Cryopreserved human hepatocytes were purchased from XenoTech LLC (Lenexa, KS), the Research Institute for Liver Disease (Shanghai, China), and In Vitro Technologies (Baltimore, MD). Just before the uptake experiment, the hepatocyte suspension was thawed at 37°C and poured into Tube A of the hepatocyte isolation kit (XenoTech LLC) containing supplemented Dulbecco's modified Eagle's medium and isotonic Percoll and then centrifuged (70g) for 5 min at 25°C. After the supernatant was removed, the cells were resuspended in 5 ml of supplemented Dulbecco's modified Eagle's medium in Tube B of the hepatocyte isolation kit. The number of viable cells was then determined using trypan blue staining. The cell viability of human hepatocytes ranged from 75 to 97%. Subsequently, the cells were resuspended in the remaining medium from Tube B (approximately 40 ml) and then centrifuged (50g) for 3 min at 25°C, followed by removal of the supernatant. Finally, the cells were resuspended in the Krebs-Henseleit buffer at a density of 2.0 × 106 viable cells/ml for the uptake experiment.
Determination of Statin Uptake Clearance Using Hepatocytes.
This experiment was performed as described previously with a minor modification (Hirano et al., 2004). Before the uptake studies, the cell suspensions were prewarmed at 37°C for 3 min. The uptake reaction was initiated by adding an equal volume of buffer-containing drugs to the hepatocyte suspension. After incubation at 37°C for 0.5, 1.5, and 2.5 min, the reaction was terminated by separating the cells from the substrate solution. For this purpose, an aliquot of 80 to 100 μl of incubation mixture was placed in a 0.4-ml centrifuge tube containing 50 μl of 5 M ammonium acetate under a 100-μl layer of oil mixture (density, 1.015, a mixture of silicone oil and mineral oil; Sigma-Aldrich), and subsequently the sample tubes were centrifuged for 15 s using a tabletop centrifuge (10,000g, MC-150; Tomy Seiko, Tokyo, Japan). During this process, hepatocytes passed through the oil layer into the aqueous solution. Tubes were frozen in liquid nitrogen immediately after centrifugation and stored at −30°C until quantification. An aliquot was taken from the upper media portion and quenched in methanol, and the cells were taken from the centrifuge tube and sonicated in a new tube, containing methanol, to disintegrate them. The samples were vortexed and centrifuged, and supernatants from both the media and cell portions were analyzed by liquid chromatography/tandem mass spectrometry (LC/MS/MS). The area under the statin concentrations in the incubation buffer (AUCbuf0–t) was calculated using a trapezoidal method. The amount of statin uptake into hepatocytes per 106 viable cells (Xhep) normalized by the buffer concentration (Cbuf) can be described by the following equation:
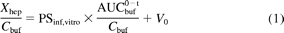 where PSinf,vitro and V0 represent uptake clearance into hepatocytes and the initial distribution volume, respectively. Based on eq. 1, the Xhep(t)/Cbuf(t) value was plotted against the AUCbuf0–t/Cbuf(t) value, and PSinf,vitro was determined as the initial slope of the plot and expressed as the in vitro uptake clearance (μl/min/106 cells). A physiological scaling factor of 1.2 × 108 cells/g liver was used for scaling up to the organ level (Iwatsubo et al., 1997).
where PSinf,vitro and V0 represent uptake clearance into hepatocytes and the initial distribution volume, respectively. Based on eq. 1, the Xhep(t)/Cbuf(t) value was plotted against the AUCbuf0–t/Cbuf(t) value, and PSinf,vitro was determined as the initial slope of the plot and expressed as the in vitro uptake clearance (μl/min/106 cells). A physiological scaling factor of 1.2 × 108 cells/g liver was used for scaling up to the organ level (Iwatsubo et al., 1997).
In Vivo Pharmacokinetic Analysis in Rats.
Male SD rats, weighing approximately 240 to 300 g, were used for these experiments. Under ether anesthesia, the femoral artery was cannulated with a polyethylene catheter (SP-31; Natsume Seisakusho Co., Tokyo, Japan) for the collection of blood samples. The bile duct was cannulated with a polyethylene catheter (PE-10; Natsume Seisakusho Co.) for bile collection, and the bladder was cannulated with a silicon catheter to collect urine. The femoral vein was cannulated with a polyethylene catheter (SP-31; Natsume Seisakusho Co.) for the administration of statins. Each rat was placed in a Bollman cage and allowed to recover from the anesthesia before the experiments were continued. The rats were given statins intravenously at 1 μmol/kg (pitavastatin and atorvastatin) or 0.5 μmol/kg (fluvastatin). Blood samples were collected at the designated times and centrifuged at 1500g for 10 min at 4°C to obtain plasma. Bile and urine samples were collected in preweighed test tubes at the designated intervals throughout the experiment. All the samples were stored at −30°C until quantification. Plasma, bile, and urine samples were deproteinized with two volumes of methanol and centrifuged at 15,000g for 10 min at 4°C. The supernatant was subjected to LC/MS/MS analysis.
Liver Perfusion Study (Multiple Indicator Dilution Method).
The procedures are basically as reported (Miyauchi et al., 1993; Akita et al., 2002). Under ether anesthesia, the portal and hepatic veins were cannulated to allow infusion of the perfusate and to allow the outflow to be collected, respectively. The perfusate consisted of 3% bovine serum albumin (BSA) in the Krebs-Ringer bicarbonate buffer, pH 7.4, and the flow rate was 30 ml/min. After the stabilization period of 10 min, 200 μl of the perfusion solution containing FD-4 (100 μM), an extracellular reference, and each statin (50 μM) was administered as a bolus into the portal vein. After administration, the total effluent from the hepatic venous vein was collected at 1-s intervals for 10 s. The concentration of FD-4 and statins in the collected samples was determined using a fluorescence plate reader (485 nm for excitation and 520 nm for emission, FluoStar Optima; BMG Labtech GmbH, Offenburg, Germany) and by LC/MS/MS, respectively. The natural logarithm of the ratio of FD-4 to statin concentration in the outflow was plotted as a function of time. The initial slope of this plot, calculated by linear regression analysis using initial four to five data points, reflects the influx rate constant (K1). The unbound uptake clearance (PSinf,MID) can be calculated by the following equation (eq. 2):
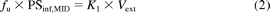 where fu and Vext represent the unbound fraction of statins in the perfusion buffer containing BSA and the extracellular volume, which can be estimated by multiplying the perfusate flow rate by the transit time of the extracellular reference, respectively.
where fu and Vext represent the unbound fraction of statins in the perfusion buffer containing BSA and the extracellular volume, which can be estimated by multiplying the perfusate flow rate by the transit time of the extracellular reference, respectively.
Determination of the Metabolic Clearance of Statins Using Liver Microsomes.
Rat liver microsomes were prepared from four rats using standard procedures and stored at −80°C until use, and human liver microsomes were purchased from XenoTech LLC. Each statin was incubated with a reaction mixture consisting of liver microsomes (final concentration, 1 mg/ml) and an NADPH-generating system (0.8 mM NADP+, 8 mM glucose 6-phosphate, 1 U/ml glucose-6-phosphate dehydrogenase, and 3 mM MgCl2) in the presence of 100 mM phosphate buffer, pH 7.4. After preincubation at 37°C for 5 min, each statin (final concentration, 0.1 μM) was added to initiate the enzyme reaction. The reaction was terminated at the following time points by mixing the reaction mixture with a 4-fold volume of methanol, followed by centrifugation at 15,000g for 10 min at 4°C. The time points when the reaction was terminated were 0, 5, 15, 30, and 60 min for the metabolic reaction of pitavastatin in rat microsomes; 0, 5, 15, 30, 60, 90, and 120 min for that in human microsomes; and 0, 5, 15, and 30 min for the metabolic reaction of atorvastatin and fluvastatin in rat and human microsomes. The metabolic reaction was continued until the fraction metabolized was greater than 15% so that we could obtain reliable parameters. The actual fractions metabolized at the end of experiment were 30, 25, and 34% (rat microsomes) and 23, 52, and 65% (human microsomes) for pitavastatin, atorvastatin, and fluvastatin, respectively.
The supernatant was subjected to LC/MS/MS analysis. The metabolic velocity was calculated as the slope of the natural log (concentration)-time plot. The in vitro intrinsic metabolic clearance (CLmet,int,vitro) was calculated by dividing initial metabolic velocity by the statin concentration in the incubation buffer corrected by the fraction unbound to liver microsomes. A physiological scaling factor of 44.8 mg protein/g liver (rats) or 48.8 mg protein/g liver (humans) was used for scaling up to the organ level (Naritomi et al., 2001).
Determination of Protein Binding.
Binding of statins to plasma proteins, liver microsomes, or perfusion buffer containing BSA used in the MID study was determined by an ultrafiltration method. Rat plasma was obtained by the centrifugation of blood from male SD rats, and human serum was purchased from Cosmo Bio Co. (Tokyo, Japan). Each statin (final concentration; 5, 0.1, and 50 μM for plasma, microsome, and perfusate, respectively) was added to the protein solution and incubated at 37°C for 5 min. The specimen was applied to YM-30 Centrifree devices (Millipore Corporation, Billerica, MA), and the devices were centrifuged at 2000g for 5 min at 37°C. The fraction unbound was calculated as concentration found in filtrate per total concentration. The concentrations of the drugs in the filtrate and the protein solution before filtration were determined by LC/MS/MS. The adsorption of pravastatin, pitavastatin, and atorvastatin to the filter was negligible, and that of fluvastatin was 19%. The binding of fluvastatin was normalized with respect to the filter blank.
Determination of the Blood-to-Plasma Concentration Ratio.
To determine the blood-to-plasma concentration ratio (RB) values, blood was obtained from male SD rats. Statins (final concentration, 1 μM) were individually added to the blood samples, and they were incubated together at 37°C for 5 min. Plasma was prepared by centrifugation of the blood samples (1500g, 5 min). The concentrations of the statins in the plasma samples were determined by LC/MS/MS. RB values in humans were cited from the previous studies (Tse et al., 1993; Lennernäs and Fager, 1997; FDA-approved package). The unbound fraction in the blood (fB) was calculated by dividing the unbound fraction in plasma by RB.
LC/MS/MS Analysis.
The appropriate standard curves were prepared in the equivalent blank matrix and used for each analysis. High-concentration samples were diluted appropriately with blank matrix. R-122798 (for pravastatin and pitavastatin) and cerivastatin (for atorvastatin and fluvastatin) were used as analytical internal standards.
The LC/MS/MS system consisted of an Alliance 2795 separations module with an autosampler (Waters, Milford, MA) and a Micromass Quattro Ultima tandem quadrupole mass spectrometer with an electron ion spray interface (Waters). The desolvation gas (nitrogen) flow rate was 650 l/h; the cone gas (nitrogen) flow rate was 30 l/h; the source temperature was 150°C; and the desolvation temperature was 450°C.
It was operated in a multiple reaction monitoring mode using negative ion mode. Deprotonated molecular ions were formed using a capillary energy of 3.2 kV and cone energies of 50 V (pravastatin), 45 V (pitavastatin and cerivastatin), and 40 V (atorvastatin and fluvastatin). Product ions formed at collision energies of 12 eV (pravastatin, m/z 423.5→321.2; cerivastatin, m/z 458.5→396.1), 12 eV (pitavastatin, m/z 420.5→358.1; R-122798, m/z 409.5→321.2), 28 eV (atorvastatin, m/z 557.6→397.2), and 15 eV (fluvastatin, 410.3→348.2) were monitored. The mobile phase used for high-performance liquid chromatography was 0.1% formic acid/acetonitrile = 73:27 (for pravastatin and pitavastatin) or 55:45 (for atorvastatin and fluvastatin), and the flow rate was 0.4 ml/min. Chromatographic separation was achieved on a C18 column (Capcell Pak C18 MG-II column, 50 × 2 mm; particle size, 3 μm; Shiseido, Tokyo, Japan).
Eight-point calibration curves were generated by plotting the peak area ratios of analyte/internal standard against the nominal analyte concentrations using linear regression with 1/(area ratio)2 weighting. The typical R-squared value of the calibration curves was 0.997 to 0.999. The concentration range was 1 to 1000 nM for atorvastatin and 3 to 3000 nM for the other statins. The back-calculated concentrations of all the calibration standards were to be within 15% of their individual nominal concentrations (±20% at the lower limit of quantitation). Intraday and interday variability for the quantification of statins was less than 15%.
Pharmacokinetic Analysis in Rats.
Pharmacokinetic parameters were calculated using noncompartmental analysis. Area under the plasma concentration-time curve (AUCP) was calculated using the trapezoidal rule with extrapolation to infinity, and total blood clearance (CLtot,B) was estimated as dose/(AUCP × RB). CLtot,B was regarded as the hepatic clearance (CLH) because the urinary excretion of all the statins in male SD rats was negligible. Hepatic availability (FH) of pitavastatin, atorvastatin, and fluvastatin was calculated from the following equation:
 where QH represents the hepatic blood flow. FH of pravastatin could not be estimated accurately from eq. 3 because CLH of pravastatin was hepatic blood flow-limited; in other words, FH was extremely small. Therefore, FH of pravastatin was obtained by dividing its bioavailability (Watanabe et al., 2009) by the fraction absorbed (Komai et al., 1992), assuming negligible metabolism in the small intestine. Overall intrinsic clearance (CLint,all,vivo) was calculated from the following equations using a dispersion model (Roberts and Rowland, 1986).
where QH represents the hepatic blood flow. FH of pravastatin could not be estimated accurately from eq. 3 because CLH of pravastatin was hepatic blood flow-limited; in other words, FH was extremely small. Therefore, FH of pravastatin was obtained by dividing its bioavailability (Watanabe et al., 2009) by the fraction absorbed (Komai et al., 1992), assuming negligible metabolism in the small intestine. Overall intrinsic clearance (CLint,all,vivo) was calculated from the following equations using a dispersion model (Roberts and Rowland, 1986).



The hepatic blood flow rate was set at 50 to 80 ml/min/kg for rats and at 17 to 25.5 ml/min/kg for humans, and DN was set at 0.17. A physiological scaling factor of 41.2 g liver/kg b.wt. (rats) or 24.1 g liver/kg b.wt. (humans) was used for scaling down to the organ level.
Pharmacokinetic Analysis in Humans.
The availability in the liver (FH) of pravastatin and atorvastatin was calculated using eq. 3 and the plasma concentration and urinary excretion data after intravenous administration in the clinical studies and fB values (Singhvi et al., 1990; FDA-approved package). In the case of fluvastatin, FH was calculated by dividing its bioavailability (0.33) by the fraction absorbed in humans (0.9) because its hepatic clearance (16 ml/min/kg) was close to the hepatic blood flow rate (Tse et al., 1992; Lindahl et al., 1996). FH of pitavastatin was obtained from the following equation (eq. 7) using the plasma concentration data after oral administration in humans (Ando et al., 2005) and fraction absorbed (Fa) in rats (0.83) (Kimata et al., 1998), assuming no interspecies differences in Fa and negligible metabolism in the small intestine:
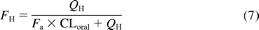 where CLoral is blood clearance after oral administration. Subsequently, CLint,all,vivo of each statin was calculated from eqs. 4 to 6.
where CLoral is blood clearance after oral administration. Subsequently, CLint,all,vivo of each statin was calculated from eqs. 4 to 6.
Results
Uptake Clearances of Statins Determined Using Freshly Isolated Rat Hepatocytes and Cryopreserved Human Hepatocytes.
The uptake clearances (PSinf,vitro) of the statins were determined using rat and human hepatocytes (Fig. 1). The uptake clearance was markedly in the presence of excess amounts of the statins in both rat and human hepatocytes. PSinf,vitro determined at 0.1 μM are scaled up to the in vivo value per unit liver weight using the following physiological scaling factors: 41.2 g liver/kg, 1.2 × 108 cells/g liver for comparison with the corresponding PSinf,MID. PSinf,vitro of statins in rats were almost similar or somewhat lower than PSinf,MID (Table 3).

Uptake clearance of statins in isolated human and rat hepatocytes. Uptake clearance of four statins by hepatocytes determined at 37°C at two concentrations (closed bar, 0.1 μM; open bar, 100 μM) by the oil filtration method. Cryopreserved human hepatocytes (three independent batches depicted as Lots A, B, and C) and freshly isolated rat hepatocytes were used in the determinations. Cells were incubated with statins for 0.5, 1.5, and 2.5 min; subsequently, reactions were terminated by rapid separation of the cells from the uptake buffer using centrifugation. The uptake is represented by the amount associated with the cell specimens divided by the statin concentrations in the uptake buffer. Data represent the mean ± S.E. (n = 3).
Determination of the in Vivo Intrinsic Hepatic Clearance of Statins in Rats.
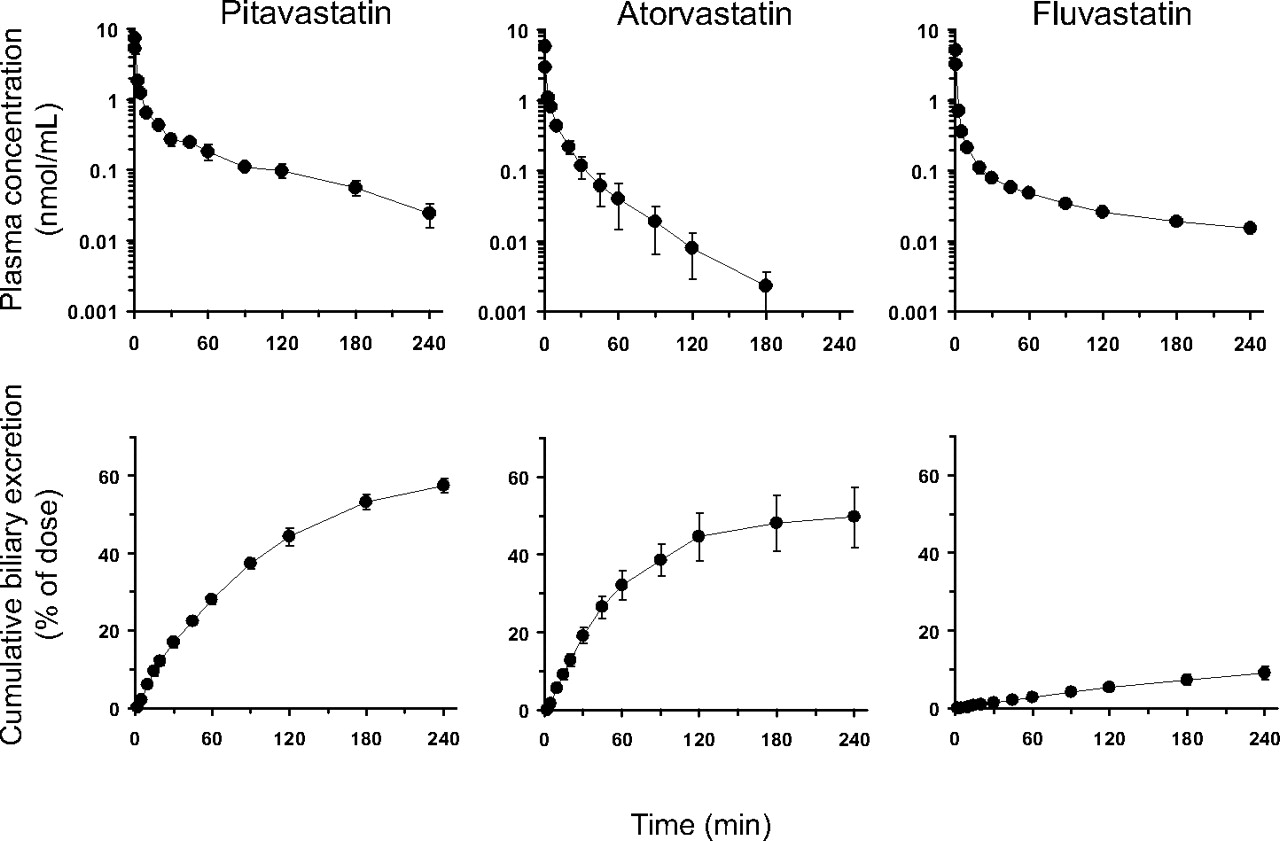
Figure 2 shows time profiles of the plasma concentrations and the cumulative amount of statins excreted into the bile after intravenous administration. Approximately 50% of the dose was recovered in the bile after the administration of pitavastatin and atorvastatin, whereas fluvastatin was slightly excreted into the bile as unchanged form. Urinary excretion of all the statins was negligible. The pharmacokinetic parameters of the statins were determined by noncompartmental analysis (Table 1). The plasma/serum unbound fraction and RB of each statin were also measured, and the findings are summarized in Table 2.

Plasma concentration-time profiles (top) and cumulative biliary excretion (bottom) of statins after intravenous administration to male SD rats. Male SD rats were given statins: pitavastatin (1 μmol/kg, left), atorvastatin (1 μmol/kg, middle), and fluvastatin (0.5 μmol/kg, right), intravenously. The plasma concentrations were determined over 240 min after administration for pitavastatin, 180 min for atorvastatin, and 240 min for fluvastatin. Bile was collected from the common bile duct via an indwelling cannula, and the cumulative amount of biliary excretion was determined. Data represent the mean ± S.E. (n = 3).
Pharmacokinetic parameters of statins in rats
RB and unbound fraction in the perfusion buffer, rat plasma, and human serum, as well as fB for each statin
Intrinsic clearances of statins related to their hepatic clearance in rats
All the intrinsic clearances are scaled up to the in vivo clearance values per unit liver weight using the following physiological scaling factors: 41.2 g liver/kg, 1.2 × 108 cells/g liver, and 44.8 mg microsomal protein/g liver. Data represent the mean ± S.E. (n = 3).
Determination of the in Situ Intrinsic Hepatic Uptake Clearance of Statins.
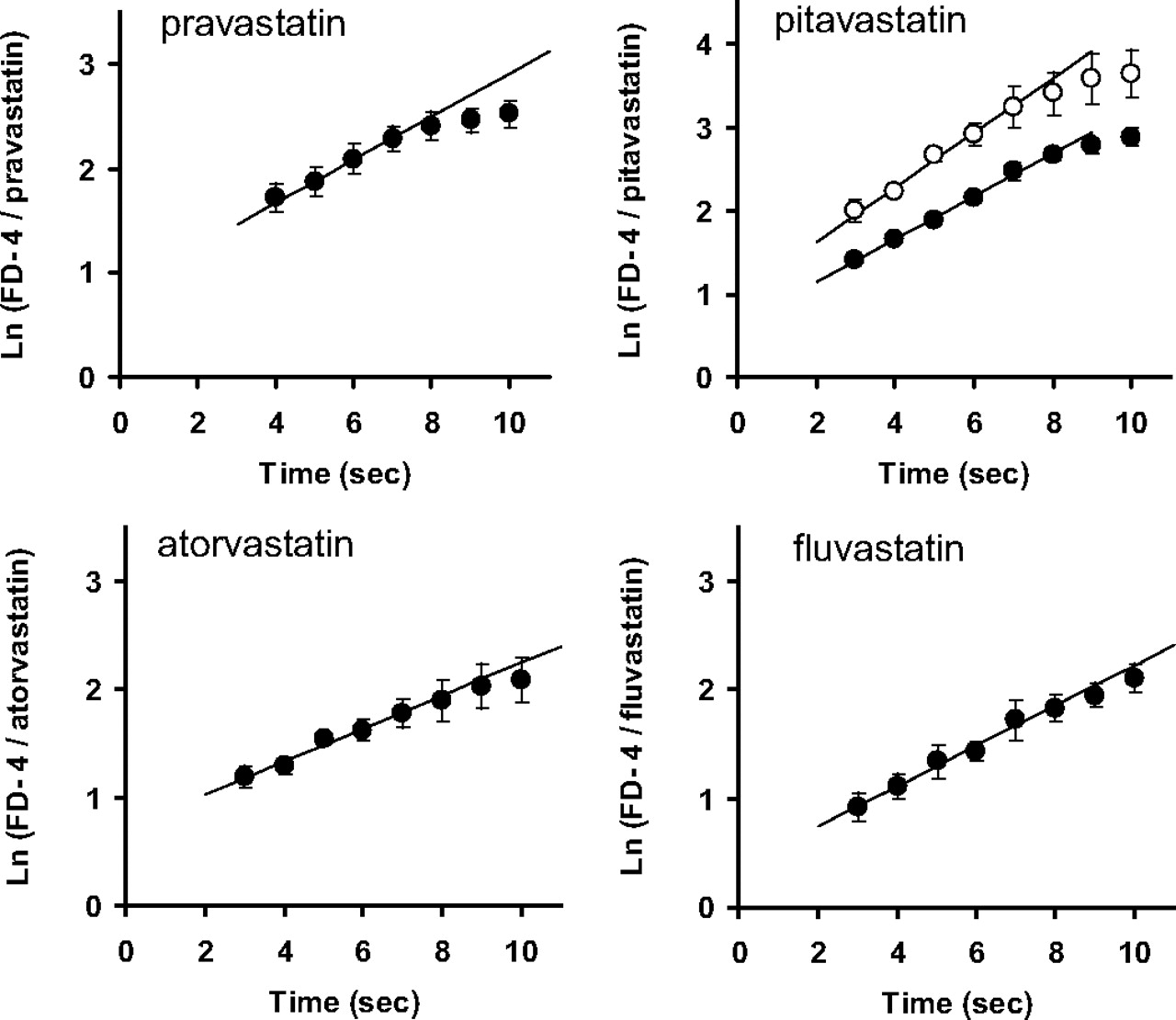
The intrinsic hepatic uptake clearance (PSinf,MID) was determined by the multiple indicator dilution method using FD-4 as an extracellular space marker. The natural logarithm of the ratio of the concentration of FD-4 to that of each statin in the outflow (ratio plot) is given as a function of time in Fig. 3. The intrinsic hepatic uptake clearance (PSinf,MID) of statins was determined from the slope of the plot and unbound fraction in the perfusate (Tables 2 and 3. For pitavastatin, to validate the unbound uptake clearance, PSinf,MID was determined using the perfusion buffers containing 1.5 or 3% BSA. The unbound fractions of pitavastatin in the presence of 1.5 and 3% BSA were 0.0859 and 0.0489, respectively, and PSinf,MID was similar [74.1 ± 25.1 and 91.5 ± 8.5 ml/min/g liver (mean ± S.E.), respectively].

Time profiles of the natural logarithm of the concentration ratio of FD-4 to statins in the outflow. After 10-min preperfusion, each statin (50 μM) and FD-4 (100 μM), an extracellular reference, were injected into the portal vein. After injection, the total effluent from the hepatic vein was collected at 1-s intervals for 10 s. For pitavastatin, the injected solution contained 3% (●) or 1.5% (○) BSA. Data represent the mean ± S.E. (n = 3).
Comparison of Intrinsic Clearances in the Hepatic Elimination of Statins by Rats.
Intrinsic clearances related to the hepatic clearance of statins, such as PSinf,MID, PSinf,vitro, CLmet,int,vitro, and CLint,all,vivo, by rats are summarized in Table 3. All the parameters were expressed as the value per unit weight of the liver. CLint,all,vivo was determined from eqs. 4 to 6 using FH and fB of each statin (Tables 1 and 2) and QH (1.21–1.94 ml/min/g liver). PSinf,MID and PSinf,vitro of statins were similar to CLint,all,vivo, whereas CLmet,int,vitro was much lower than CLint,all,vivo (Fig. 4).

Comparison of the hepatic overall intrinsic clearance of statins with the hepatic uptake clearance or metabolic clearance. In vivo hepatic overall intrinsic clearances of statins in rats and humans are plotted against the uptake clearance or metabolic clearance. ●, uptake clearance in humans predicted using scaling factor (Table 4); ○, metabolic clearance determined using human liver microsomes (Table 4); ■, uptake clearance in rats determined using a MID method (Table 3); □, metabolic clearance determined using rat liver microsomes (Table 3). The straight line indicates a 1:1 correlation. Each point represents the mean ± S.E. (n = 3).
Intrinsic clearances regarding the hepatic clearance of statins in humans
All the intrinsic clearances are scaled up to the in vivo clearance values per unit liver weight using the following physiological scaling factors: 24.1 g liver/kg, 1.2 × 108 cells/g liver, and 48.8 mg microsomal protein/g liver. Data represent the mean ± S.E. (n = 3).
Comparison of Intrinsic Clearances in the Hepatic Clearance of Statins by Humans.
Intrinsic clearances of statins by humans are summarized in Table 4. All the parameters are expressed as the value per the unit weight of the liver except for the scaling factor. The uptake clearances determined by the in vitro model were extrapolated to in vivo clearances using physiological and drug-related scaling factors, and the latter scaling factor was defined as the ratio of the in situ to in vitro uptake clearances for each statin in rats. As observed in rats, the predicted uptake clearances (PSinf,vivo,predicted) were similar to CLint,all,vivo obtained from the clinical studies. In contrast, the CLmet,int,vitro of atorvastatin and fluvastatin was markedly low to account for CLint,all,vivo (Fig. 4), although these statins are mainly eliminated from the liver through metabolism by P450.
Discussion
In a previous study, based on pharmacokinetic analyses, we proposed that the uptake is the rate-determining process in the overall hepatic elimination of pravastatin (Watanabe et al., 2009). The present study examined the rate-determining step in the overall hepatic elimination of other statins with different elimination mechanisms: biliary excretion for pitavastatin and P450-mediated metabolism for atorvastatin and fluvastatin.
Consistent with previous studies (Hirano et al., 2004), the uptake of pitavastatin by human hepatocytes was saturable (Fig. 1). In addition, uptake of atorvastatin and fluvastatin was also saturable in human hepatocytes (Fig. 1). Saturable uptake of atorvastatin is in good agreement with the clinical report in which coadministration of rifampicin, an inhibitor of OATP1B1, greatly enhanced the systemic exposure of atorvastatin, and the systemic exposure of atorvastatin is affected by the genotypes of OATP1B1 (Lau et al., 2007; He et al., 2009). Although an in vitro study using cDNA transfectants showed that fluvastatin is a substrate of OATP1B1 (Kopplow et al., 2005; Noé et al., 2007), the systemic exposure of fluvastatin was independent of the OATP1B1 genotypes (Niemi et al., 2006), suggesting that a transporter distinct from OATP1B1 may play a major role in the hepatic uptake of fluvastatin. An in vitro study using cDNA transfectants showed that fluvastatin is also a substrate of other hepatic uptake transporters, such as OATP1B3 and OATP2B1 (Kopplow et al., 2005; Noé et al., 2007). As observed for other statins, variation of the uptake transport activities has a reciprocal relationship to the blood concentration of fluvastatin, and particularly, reduction of the uptake activity will increase the risk of an adverse reaction. Because of its negligible urinary excretion of fluvastatin, the variation in its hepatic uptake will have only a minimal impact on the liver concentration (Watanabe et al., 2009) and pharmacological response.
The rate-determining process was identified in rats by comparing the in vivo intrinsic hepatic clearance and uptake clearance. To obtain intrinsic hepatic clearance, the pharmacokinetics of pitavastatin, atorvastatin, and fluvastatin was examined in rats. Considering the recovery of the unchanged forms in the bile, biliary excretion and metabolism make a similar contribution to the intrinsic hepatic clearance of pitavastatin and atorvastatin, whereas hepatic metabolism is the predominant pathway for elimination of fluvastatin in rats. The uptake clearance (PSinf,MID) of the statins was found to be similar to their corresponding intrinsic hepatic clearances (CLint,all,vivo) (Table 3; Fig. 4). Namely, uptake is the rate-determining process in the hepatic elimination of the statins. In contrast, intrinsic sequestration clearances (metabolism for fluvastatin, biliary excretion and metabolism for atorvastatin and pitavastatin) were negligibly low accounting for the CLint,all,vivo (Table 3; Fig. 4). This poor predictability is likely the result of active uptake from the blood in the sinusoidal membrane.
The rate-determining process in the hepatic elimination of the statins was also examined in humans. In a previous study, we introduced the rat scaling factor to extrapolate in vitro uptake clearance of pravastatin determined using human hepatocytes to the in vivo clearance, which provided reasonable parameters to reproduce the plasma concentration-time profiles of pravastatin after intravenous and oral administration (Singhvi et al., 1990; Watanabe et al., 2009). In this study, we determined the scaling factor for each statin in rats by comparing their in situ and in vitro uptake clearances. The scaling factors of statins appear to be compound-dependent, ranging from 1.7 to 4.5 (Table 4). The scaling factor of the statins (pravastatin, atorvastatin, and pitavastatin), the hepatic uptake of which is mediated mainly by OATP1B1, is roughly 2, whereas that of fluvastatin, the hepatic uptake of which is mediated by the transporter distinct from OATP1B1, is 2-fold greater. Therefore, it can be speculated that the scaling factor is transporter-dependent. To support this speculation, accumulation of in vitro-in vivo extrapolation data for the transporters is absolutely essential. Because 1) the in vivo uptake clearance predicted for humans was in the range of CLint,all,vivo (Table 4) and 2) CLmet,int,vitro of atorvastatin and fluvastatin determined using human liver microsomes was less than CLint,all,vivo, uptake is the most likely rate-determining process in the hepatic elimination of the statins in humans. Lau et al. (2007) also suggested that hepatic uptake was important for systemic exposure of atorvastatin based on the clinical drug-drug interaction study between atorvastatin and a potent inhibitor of OATPs, rifampicin. Thus, impact of the variation of the sequestration clearance (metabolism or biliary excretion) caused by drug-drug interactions or genetic polymorphisms depends on the rate-determining process and will be smaller for these statins compared with drugs that achieve a rapid equilibrium. Indeed, the increase (2.5–3-fold) in the AUC of atorvastatin caused by concomitant use of itraconazole, a potent CYP3A4 inhibitor, was less remarkable than for other CYP3A4 substrates, such as midazolam and triazolam (5–10-fold increase) (Venkatakrishnan et al., 2000; Shitara and Sugiyama, 2006). BCRP genotypes produce no significant interindividual variation of the systemic exposure of pitavastatin (Ieiri et al., 2007), although they play a predominant role in mice (Hirano et al., 2005). It should be noted that, irrespective of the rate-determining process, the sequestration clearance is the predominant factor determining the liver concentration of the statins (Watanabe et al., 2009). Thus, inhibition of CYP3A4 or BCRP polymorphisms results in a significant increase in the liver concentration of atorvastatin and pitavastatin, respectively, leading to the enhancement of their pharmacological action. To validate the prediction of the rate-determining process of these statins, information regarding the tissue concentration-time profile is necessary. Clinical studies using positron emission tomography/single photon emission computed tomography will allow an advancement to improve the predictability of pharmacokinetic parameters.
To determine the intrinsic hepatic clearance that comprises uptake, sinusoidal efflux, and metabolism, Soars et al. (2007) proposed a “media loss” assay using isolated hepatocytes. Using this method, an intrinsic clearance can be determined from the concentration-time profile of drugs in incubation media and the initial amount of drugs applied. In theory, the method must be able to provide a reliable overall hepatic intrinsic clearance. However, as described in the report by Soars et al. (2007), this method considerably underestimates the in vivo overall intrinsic clearance (with an average 16-fold error), possibly because of reduced activities of transporters and/or enzymes during the relatively long-term incubation. Therefore, at present, separate determination of the uptake and metabolic clearances will provide more reliable parameters to predict the intrinsic hepatic clearance using rat scaling factors.
The present study found that the underestimation of in vivo intrinsic hepatic clearance of the statins in the in vitro-in vivo extrapolation of metabolic clearance is because of active transport in the uptake process. Kinetic analyses showed that uptake is the rate-determining process in the hepatic elimination of the statins in rats, which likely holds true in humans. In vitro-in vivo extrapolation of the uptake clearance using a human hepatocyte model and scaling factors determined in rats should be effective for predicting in vivo intrinsic hepatic clearance of drugs when transporter(s) are involved in the hepatic uptake.
Acknowledgments.
We thank Toichiro Yamada and Ryoko Asaki for excellent technical assistance.
Footnotes
-
This work was supported by a research grant (Development of Technology to Create Research Model Cells) from the New Energy and Industrial Technology Development Organization of Japan; and Health and Labour Sciences Research Grants for Research on Regulatory Science of Pharmaceuticals and Medical Devices from Ministry of Health, Labour and Welfare, Japan.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.030254.
-
- OATP
- organic anion-transporting polypeptide
- P450
- cytochrome P450
- statin
- HMG-CoA reductase inhibitor
- BCRP
- breast cancer resistance protein
- MID
- multiple indicator dilution
- R-122798
- (3R,5R)-3,5-dihydroxy-7-[(1S2S,6S,8S,8aR)-6-hydroxy-8-(isobutyryloxy)-2-methyl-1,2,6,7,8,8a-hexahydronaphthalen-1-yl]heptanoic acid
- FD-4
- fluorescein isothiocyanate dextran 4000
- SD
- Sprague-Dawley
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- AUCbuf0–t
- area under the statin concentrations in the incubation buffer
- Xhep
- amount of statin uptake into hepatocytes per 106 viable cells
- Cbuf
- buffer concentration
- BSA
- bovine serum albumin
- K1
- influx rate constant
- PSinf,MID
- unbound uptake clearance
- RB
- blood-to-plasma concentration ratio
- fB
- unbound fraction in blood
- AUCP
- area under the plasma concentration-time curve
- CLtot,B
- total blood clearance
- CLH
- hepatic clearance
- FH
- hepatic availability
- CLint,all
- overall intrinsic clearance
- Fa
- fraction absorbed.
- Received September 16, 2009.
- Accepted October 23, 2009.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}