


Abstract
It is important to gain an understanding of the pharmacological activities of metabolite(s) of compounds in development, especially if they are found in systemic circulation in humans. Pharmacological evaluation of metabolites is normally conducted with synthetic standards, which become available during various stages of drug development. However, the synthesis of metabolite standards may be protracted, taking anywhere from several weeks to months to be completed. This often slows down early pharmacological evaluation of metabolites. Once a metabolite(s) is found to possess comparable (or greater) pharmacological activity than the parent compound, additional studies are performed to better understand the implications of circulating pharmacologically active metabolite(s). To conduct some of these studies as early as possible without slowing the progression of a compound in development is important, especially if critical go or no-go decisions impinge on the outcomes from these studies. Early pharmacological evaluation of significant metabolites is hereby proposed to be conducted in the drug discovery stage so that all pertinent studies and information can be gathered in a timely manner for decision-making. It is suggested that these major metabolites be isolated, either from biological or chemical sources, and quantified appropriately. For biologically generated metabolites, NMR is proposed as the tool of choice to quantitate these metabolites before their evaluation in pharmacological assays. For metabolites that have the same UV characteristics as the parent compound, quantitation can be conducted using UV spectroscopy instead of NMR. In this article, we propose a strategy that could be used to determine the pharmacological activities of metabolites isolated in submilligram quantities.
Introduction
Pharmacological evaluation of new chemical entities is traditionally performed in early drug discovery. Enzyme and cell-based assays are often used to initially assess the potential pharmacological activities of compounds before further evaluation in animal models. Frequently, the relationship between the structure and pharmacological activity is further studied by chemically synthesizing close structural analogs with the intention of optimizing drug-receptor interaction for improved potency. This structure-activity relationship work is critical during the lead optimization phase of new potential therapeutic agents because modifications of chemical templates can also lead to significant alterations in physicochemical properties, which in turn can dramatically influence the absorption, distribution, metabolism, and excretion characteristics of compounds. Significant chemical synthesis is done in the lead optimization stage to generate compounds with desired pharmacological and absorption, distribution, metabolism, and excretion properties. An alternative, yet less exploited, way of creating close analogs from a pharmacologically active template is through biological means, such as enzyme-mediated metabolism or microbial conversion.
During early drug discovery studies, metabolic modifications of chemical templates by various subcellular and cellular systems from different animal species are routinely investigated. Metabolite profiles, which are often obtained by combination of liquid chromatography (LC), mass spectrometry (MS), and UV spectroscopy, identify new chemical entities produced by these biological enzymes. However, because of the low levels of these metabolites, further pharmacological evaluations of these compounds have not routinely been performed in the past.
Metabolic conversion has not traditionally been considered as a source for generating new chemical entities for further pharmacological evaluation. In addition to regarding metabolites merely as excretory products, there are several other reasons that medicinal chemists have not considered metabolites as potential drug candidates. In the past, elucidating the structures of these metabolites was a challenge. Providing a list of hypothetical structures based on inconclusive mass spectral data was one of the reasons for lack of interest in these metabolites by the medicinal chemists and biologists. However, with advancements in mass spectrometry and NMR techniques, it has become a routine practice to obtain structures of metabolites in early discovery (Mutlib and Shockcor, 2003). A significant hurdle in the past has been the inability to isolate sufficient quantities of metabolites in relatively pure form for further evaluation. Recently, it has become easier to produce sufficient amounts of metabolites using one of the various biological or biomimetic systems. It has been shown that these biological systems can be used reliably to generate metabolites in the microgram to milligram range. Despite the ability to generate and isolate sufficient quantities of pure samples of metabolites, the challenge has been to obtain an accurate weight of the compounds for proper evaluation in pharmacological assays.
A strategy advocating the use of NMR as a quantitative tool to determine the concentrations of biologically synthesized metabolites has been discussed in the literature and elsewhere (Espina et al., 2009). Biologically isolated metabolites have not traditionally been considered as a source to generate reference standards for further quantitative work. However, because of the U.S. Food and Drug Administration guidance on safety testing of metabolites, some laboratories have contemplated using these biologically synthesized metabolites as “reference standards” for preliminary quantitative studies. 1H NMR has been proposed as the method of choice for authenticating and quantitating these biologically synthesized metabolites before their further use as standards in more sensitive LC-MS-based quantitative assays (Vishwanathan et al., 2009). We have successfully used this approach to obtain metabolite concentrations and exposure values using sample pooling methods (Vishwanathan et al., 2009). A secondary consequence of being able to obtain accurate weights of isolated metabolites is further evaluation of their pharmacological activities. Once the exact structure and concentration of the isolated metabolite is known, it can be submitted to pharmacologists for further testing in enzyme- and cell-based assays. In this article, we describe a strategy that was used to effectively elucidate the structure and determine the concentrations of metabolites before evaluation of pharmacological activities of these biologically generated compounds. A compound that was found to be highly metabolized by human liver microsomes (Fig. 1) was studied extensively along with its major metabolite(s) in animals to better understand their pharmacokinetic behaviors in animals after it was shown that the metabolite was equipotent to the parent compound.

Metabolism of compound 1 to metabolite M1 in the presence of monkey and human liver microsomes.
Materials and Methods
Chemicals and Supplies.
Compound 1 (Fig. 1) was obtained from the Department of Chemical Sciences, Pfizer Global Research and Development, Pearl River, NY. The chemical purity of this compound was shown to be greater than 98% by HPLC-UV. NADPH was purchased from Sigma-Aldrich (St. Louis, MO). Liver microsomes from rats, cynomolgus monkeys, and humans were purchased from XenoTech, LLC (Lenexa, KS). Discovery DSC-18 (C18) cartridges were purchased from Sigma-Aldrich.
Solvents used for chromatographic analysis were HPLC or ACS reagent grade and were purchased from EMD Chemicals (Gibbstown, NJ). All deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). All general solvents and reagents were the highest grade available commercially.
In Vitro Incubations and Isolation of Metabolites for NMR Characterization and Quantitation.
A large-scale incubation of compound 1, at a concentration of 0.02 mM, with human liver microsomes was conducted to generate metabolite M1 (Fig. 1). A total of 50 2-ml incubations were conducted in 0.1 M potassium phosphate buffer (pH 7.4), containing 1 mg/ml microsomal protein, 5 mM NADPH, and 8 mM MgCl2 at 37°C for 60 min. Protein precipitation was conducted using 4 ml of 2% formic acid in acetonitrile per incubation. The tubes were vortexed for 1 min and centrifuged at 2400g for 10 min at 4°C. The supernatants were transferred to clean culture tubes after which the organic phase was removed under a stream of nitrogen at 30°C. The aqueous phase from each tube was pooled and subjected to a cleanup on solid-phase C18 extraction cartridges.
Two Discovery DSC-18 (C18, 60-ml capacity) cartridges were used for sample cleanup before further purification on a HPLC system. The cartridges were washed with 60 ml of acetonitrile followed by the same volume of H2O. The aqueous supernatant was loaded onto the cartridges (approximately 50 ml/cartridge) and allowed to flow through completely under gravity. The cartridges were then washed with 1 volume of H2O. The wash fractions were analyzed by injecting 50 μl of samples on a Thermo LTQ ion trap mass spectrometer and were found to be void of both compound 1 and M1. Elution of M1 was done using 25-ml aliquots of solvent varying in concentrations of acetonitrile in water (10–100% acetonitrile, with 10% stepwise increase). Aliquots (50 μl) of each fraction were injected on the Thermo LTQ mass spectrometer; M1 was found to elute completely in the 30, 40, and 50% fractions. These fractions were pooled (75 ml total) and subsequently reduced in volume to ∼4 ml, which was subjected to further purification on an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a fraction collector. Aliquots (500 μl) of the sample were injected on a Varian Pursuit C18 (250 × 10 mm, 5 μm; Varian, Inc., Palo Alto, CA) semipreparative column coupled to the HPLC system. The mobile phase consisted of 0.1% formic acid in H2O (A) and acetonitrile (B), which was delivered at 5 ml/min. The gradient consisted of increasing percent organic (B) from the initial value of 25 to 50% in 7 min followed by a linear ramp to 95% B in 1 min before re-equilibrating the column. Compound-related components were monitored using a UV detector set at 300 nm. Fractions containing M1 were pooled and lyophilized. Approximately 1 mg of M1 was isolated as its formate salt in >95% purity as determined by LC-MS-UV and NMR analysis. The isolated metabolite was handled in a glove box to minimize contamination with water before NMR analysis.
In Vitro Metabolism of Metabolites M1a-1 and M1a-2 (Enantiomers of M1).
Metabolites M1a-1 and M1a-2 (Fig. 2), chemically synthesized enantiomers of M1, were incubated with male Sprague-Dawley rats, male cynomolgus monkeys, and male human liver microsomes (1 mg/ml) fortified with cytosol (2 mg/ml) in the presence of NADPH (5 mM), MgCl2 (8 mM), GSH (2 mM), and UDP-glucuronic acid (2 mM) in a total volume of 1 ml (adjusted with 0.1 M phosphate buffer, pH 7.4) at 37°C for 60 min. Protein precipitation was performed using 2× volumes (2 ml) of 2% formic acid in acetonitrile, and samples were centrifuged (3400 rpm for 5 min) before transferring the supernatants to clean culture tubes. The solvents were completely removed under a steam of nitrogen at 30°C. The dried samples were reconstituted in 200 μl of the HPLC mobile phase (1:9 v/v, acetonitrile-5 mM ammonium acetate). Aliquots of reconstituted samples were analyzed by LC-MS to assess the extent of metabolism of each enantiomer in each species.
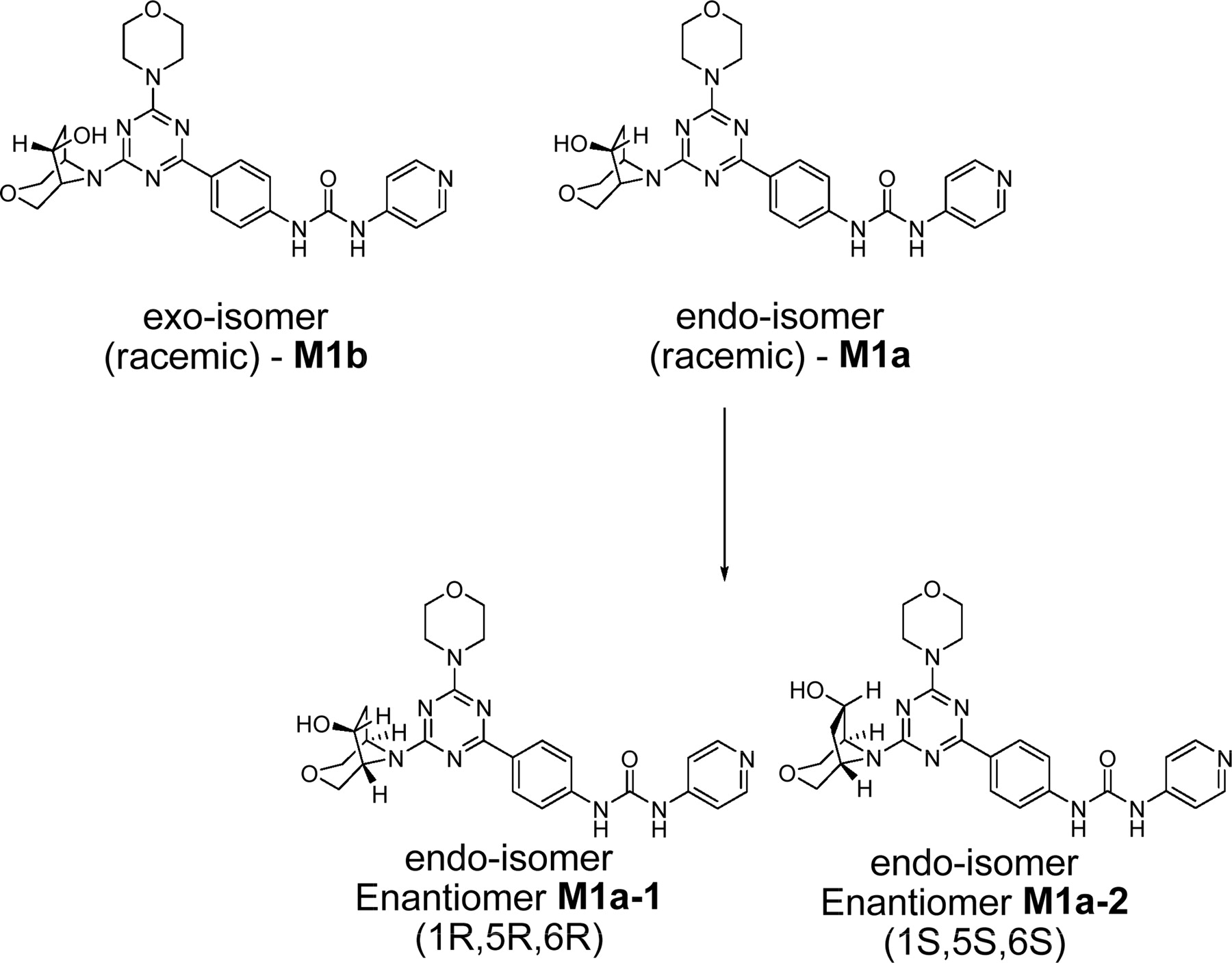
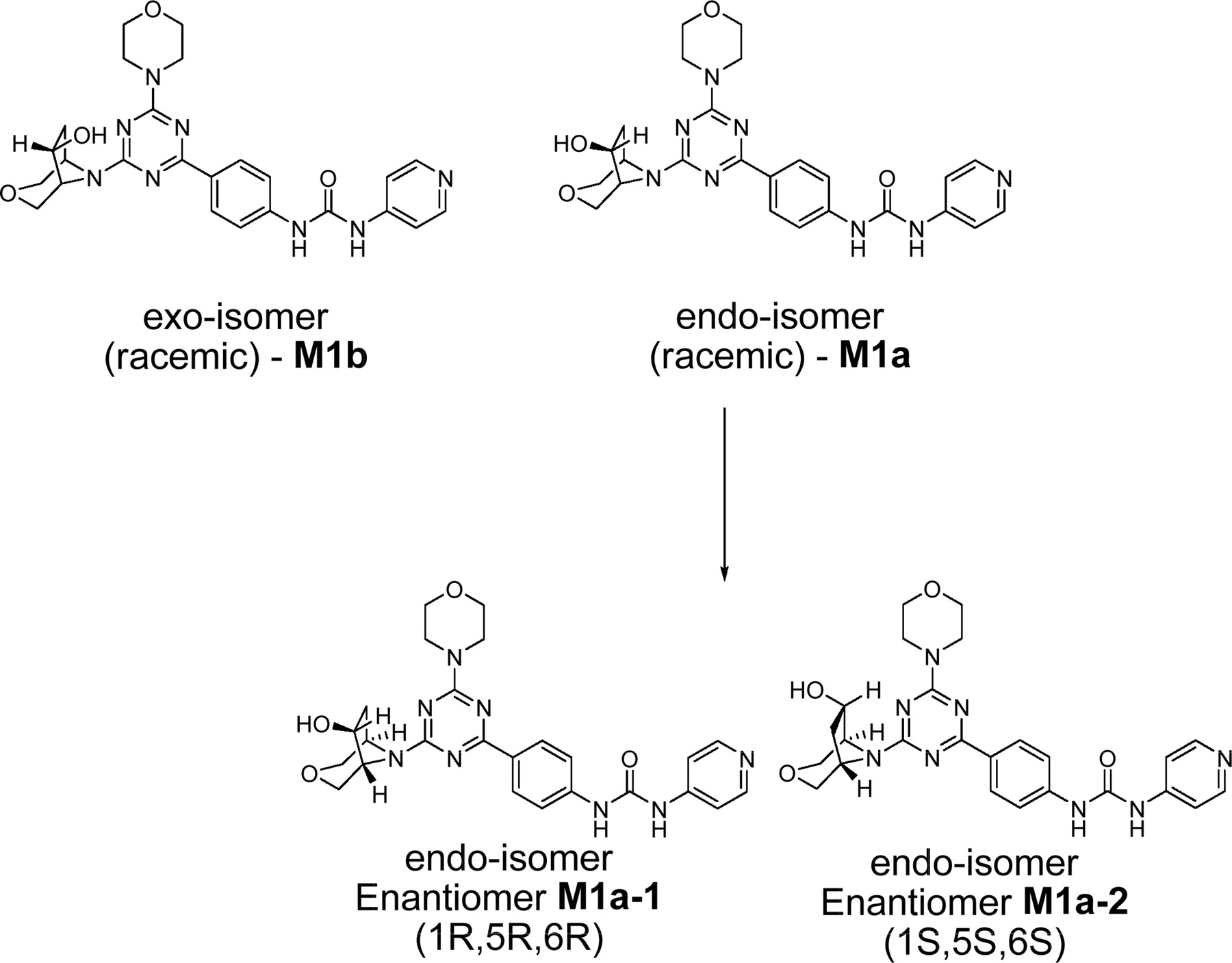
Chemical structures of synthetic isomeric forms of metabolite M1. The correct stereochemistry of metabolite M1 was found to correspond to the endo-isomer, M1a-1.
Liquid Chromatography-Mass Spectrometry.
Analysis of solid-phase extraction fractions, purity check, and metabolite characterization.
The fractions from the C18 cartridges were analyzed by flow injection analysis, whereby aliquots (50 μl) from various fractions from solid-phase extraction were injected directly onto the mass spectrometer using a 1:1 (v/v) mixture of acetonitrile-0.1% formic acid with the solvent delivered at 0.2 ml/min. Upon the isolation of the metabolite M1, LC-MS was performed to obtain structural information as well as to ensure the purity of the isolated metabolite. HPLC was conducted on a Phenomenex C18 Luna column (150 × 2.1 mm; Phenomenex, Torrance, CA) using acetonitrile and 5 mM ammonium acetate as the mobile phase. The composition of the mobile phase was changed from 10% acetonitrile to 40% in the first 18 min, after which the column was washed with 95% acetonitrile for 5 min. Subsequently, the column was re-equilibrated with 10% acetonitrile before the next injection. Deuterated water (5 mM ammonium acetate) was substituted in LC-MS experiments designed to determine the number of exchangeable protons in metabolite M1. The flow rate was set at 200 μl/min. Mass spectral data were obtained with a LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA), which was equipped with an electrospray ionization source and operated in the positive ionization mode with the electrospray needle kept at +4500 kV. Settings for the mass spectrometer were optimized to provide a structurally relevant range of product ions from MS/MS (under high-energy collisional dissociated mode) and MSn (ion trap mode) experiments. The capillary temperature and voltage were set at 370°C and 29 V, respectively. Ultrapure nitrogen was used as the nebulizer gas and curtain gas. The relative collision energy was kept at 30 eV for generating product ions in both MS/MS and MSn experiments. Other parameter settings for the analyses included arbitrary values of 55, 20, and 10 for sheath, auxiliary, and sweep gases (ultrapure nitrogen), respectively. To establish the structure of M1 unequivocally, synthetic standards of various isomeric forms of this metabolite, M1a (endo-isomer, racemic), M1a-1 (endo-isomer, enantiomer 1, 1R, 5R, 6R), M1a-2 (endo-isomer, enantiomer 2, 1S, 5S, 6S), and M1b (exo-isomer, racemic) (Fig. 2), were injected onto the LC-MS system to compare the retention times and mass spectral fragmentation patterns. Chiral separation of M1a-1 and M1a-2 was conducted as described previously (Chen et al., 2010). An aliquot of M1 was injected on the same chiral column to compare the retention time of this metabolite with the HPLC-separated enantiomers, M1a-1 and M1a-2. It was found that M1a-1 and M1a-2 had retention times of 10.2 and 13.4 min, respectively. Metabolite M1 showed a retention time identical to that of M1a-1 (Chen et al., 2010).
LC-MS analysis of microsomal and rat plasma extracts.
The microsomal and plasma extracts were subjected to LC-MS analysis using the same experimental conditions as described above.
Quantitative LC/MS analysis of plasma samples from pharmacokinetic studies.
Compound 1, M1a-1, and M1a-2 were quantified in male rat plasma using a nonvalidated LC-MS/MS assay. The assay limit of quantification was 1 ng/ml using 50-μl aliquots of rat plasma. The standard curve ranged from 1 to 1000 ng/ml for all compounds. All samples, when needed, were appropriately diluted. The analysis was accepted if three-fourths or more of the back-calculated concentrations of the calibration standards and at least two-thirds of the quality control samples were within 20% of their theoretical values. All calibration standards and quality control (QC) samples met these criteria. The coefficient of determination (r2) was >0.9991. Experimental and calibration samples were prepared by adding 50-μl aliquots of samples or standards into the appropriate well of a 96-well disposable plate. Control blanks were prepared by adding 50 μl of control matrix to the appropriate well(s). Calibration samples were prepared by adding 10 μl of the appropriate working calibration standards (prepared in acetonitrile) to the appropriate well containing 50 μl of blank plasma. To experimental and control blank samples was added 10 μl of acetonitrile. Compound 1, M1a-1, and M1a-2 were extracted from plasma by protein precipitation using 200 μl of acetonitrile containing 100 ng/ml internal standard. Samples were vortexed for 3 min and then centrifuged at 3400 rpm for 5 min. A 150-μl aliquot of the supernatant was transferred to a microplate using a Tomtec Quadra 96. Ten microliters of this sample was injected for analysis.
Chromatographic resolution was achieved on a Discovery C18 HPLC column (50 × 2.1 mm, 5 μm) at ambient temperature with the same mobile phase gradient used for the metabolite characterization (see above) All analyses were carried out using the Cohesive Aria LX2 HPLC system (Thermo Fisher Scientific) with a LEAP autosampler (LEAP Technologies, Carrboro, NC) interfaced with a Sciex API 4000 mass spectrometer (AB Sciex, Foster City, CA). The LC eluate was sprayed into the mass spectrometer at +5 kV, and desolvation of droplets was aided by heated desolvation gas. Quantitative LC-MS analysis was conducted by operating the mass spectrometer in the multiple reaction monitoring mode. The protonated molecule of the analytes (compound 1, m/z 489.2; M1, M1a-1, and M1a-2, m/z 505.2) and internal standard (m/z 475.2) were selected by Q1 set at a resolution of 0.7 Da (full width at half-maximum). Collision-activated dissociation was carried out in Q2 with nitrogen at a collision energy of 45 eV. Fragment ions of the analytes (m/z 489.2 → 395.2 and m/z 505.2 → 411.2) and internal standard (m/z 475.2 → 381.2) were mass analyzed by Q3 at a resolution of 0.7 Da (full width at half-maximum). Data acquisition and reduction including peak integration were performed with Analyst software (version 1.4.1; AB Sciex). Drug plasma concentrations were quantified using Watson LIMS (version 7.3.1; Thermo Fisher Scientific).
NMR Spectroscopy.
NMR instrument.
NMR data were recorded at 30°C on a 600-MHz Bruker Avance III spectrometer with a 5-mm CPTCI CryoProbe (Bruker BioSpin, Billerica, MA) at the basic frequency of 600.13 MHz. The spectrometer was controlled using TopSpin (version 2.1, pl1; Bruker BioSpin). All data collection was performed without spinning the samples. The probe was tuned and matched to the specific frequency using the automatic tuning method from Bruker BioSpin. The samples were field frequency locked using the 2H signal of the deuterated solvent. Shimming was performed on each sample before data acquisition using the TopShim automatic shimming method from Bruker BioSpin on the solvent peak. Each data set was collected under automation with the same parameter set using ICON-NMR software (version 3.7; Bruker BioSpin) and a SampleJet sample changer (Bruker BioSpin) or manual sample changes.
Qualitative NMR analysis of metabolite M1.
The isolated metabolite, M1, was evaporated to dryness under a nitrogen stream, dissolved in 500 μl of DMSO-d6 (D, 99.96%) (Cambridge Isotope Laboratories, Inc.), and transferred into a 5-mm NMR tube. Proton, HSQC, and heteronuclear multiple-bond correlation spectroscopy experiments were performed. All chemical shifts were referenced to the DMSO signal at 1H δ 2.49 ppm. The proton spectra were acquired with 32,768 data points over a 6602-Hz spectral window using a 30° pulse and standard pulse sequence. Data were Fourier transformed with a 0.3-Hz line broadening window function. The two-dimensional 1H-13C HSQC spectra were acquired with 1024 data points in F2 and 256 increments in F1, with 32 scans per increment, using a phase-sensitive pulse sequence.
Quantitative NMR analysis of metabolite M1.
The quantitative NMR technique used in this study was similar to what has been described previously (Espina et al., 2009). In brief, each sample was dissolved in the same volume (500 μl) of the solvent and was placed in the magnet at the same depth. Each proton spectrum was acquired with 65,536 data points over a 12,019-Hz spectral window using a 90° (8.3 μs) pulse and the gradient one-dimensional nuclear Overhauser effect spectroscopy presat pulse sequence (noesygppr1d), using a 5-mm CPTCI CryoProbe (Bruker BioSpin). Eight scans with an acquisition time of 2.7 s were collected for each spectrum. A preacquisition delay of 30 s was used during the data acquisition. The receiver gain was set automatically using the more concentrated solution and was kept constant throughout each of the data sets. Data were Fourier-transformed with a 0.3-Hz line broadening window function. The spectra were transferred into ACD/SpecManager (version 11.0; ACD/Labs, Inc., Toronto, ON, Canada). The FID Reconstruction method was applied to the full spectrum during automatic baseline correction before integration. All of the spectra in a series (absolute intensity) were combined using the SpecManager Series command. Integration of the spectra in a series was performed manually using group treatment within SpecManager. For quantitation purposes, integration of all aromatic protons (δ 8.37, 8.30, 7.55, and 7.44) of compound 1 was used to generate calibration curve as described previously (Espina et al., 2009). An average of the normalized integral value for compound 1 was plotted against the concentration values to create a calibration curve as described before (Espina et al., 2009). The table of common integrals for the series was transferred to Microsoft Excel for analysis. All samples were analyzed by NMR in triplicate (three separate determinations of the same sample on different occasions). The concentrations of QC samples and metabolite M1 were obtained using the calibration curve generated from compound 1. All spectra were referenced to the residual solvent signal.
Pharmacological Evaluation of Metabolite M1.
The exact concentration of the isolated metabolite was measured with NMR using the calibration curve constructed with compound 1 as described previously by Espina et al. (2009). In brief, the NMR spectra of the parent compound were obtained at five different concentrations and a calibration curve was created on the basis of the integration of aromatic proton signals (average). The concentration of metabolite M1 was calculated using the standard curve as described previously (Espina et al., 2009). Another metabolite of compound 1, available as a synthetic standard, was used as a quality control to ensure that the NMR quantitation was verified by gravimetric determination. The results are presented in Table 1. It was shown by Espina et al. (2009) during the validation of the quantitative NMR method that the concentrations of metabolites measured using this approach were within 10% of the gravimetrically determined nominal values. Hence, the measured concentration of the isolated metabolite was used to obtain the absolute amount of metabolite that was isolated from the biological source. Subsequently, this sample (with the concentration of the metabolite known) was used to prepare a series of diluted samples that were used for measuring the binding and functional potency of the isolated metabolite on the target receptors. In the same study, a solution of compound 1 was prepared at a concentration similar to that of the isolated metabolite M1. The concentrations of both samples were confirmed by quantitative NMR before being simultaneously evaluated in the pharmacological assays. Upon the availability of the synthetic standards of metabolite M1, pharmacological evaluations were conducted using solutions prepared from compounds that were weighed accurately using analytical balances. The pharmacological activities of various compounds, including that of the isolated metabolite M1, are shown in Table 2.
NMR determination of the concentration of M1 (isolated from microsomal incubations) produced from compound 1
Pharmacological evaluation of compound 1, isolated metabolite M1, and the synthetic enantiomers, M1a-1 and M1a-2
Metabolite M1 and M1a-1 were shown to be identical by chiral HPLC (Chen et al., 2010). For description of the pharmacological assays, please see Venkatesan et al (2010).
Pharmacokinetics of Compound 1, M1a-1, and M1a-2.
Single-dose intravenous (2 mg/kg) studies were conducted in male Sprague-Dawley rats with compounds 1, M1a-1, and M1a-2 (pure enantiomers). Male Sprague-Dawley rats (n = 3/dose group) were purchased from Charles River Laboratories, Inc. (Wilmington, MA) and acclimated in an Institutional Animal Care and Use Committee-accredited facility for a few days before dosing. Animals used in the study were surgically cannulated for jugular and femoral veins by the vendor. The compounds were formulated in 5% dextrose and 0.5% lactic acid, pH 3.5, for intravenous administration. The intravenous dose volume was 1 ml/kg. The compound was administered to animals that had access to food and water ad libitum. Serial samples (n = 10) of blood (300 μl) were taken at time points 0, 2, 10, 15, 30, 60, 120, 240, 360, and 1440 min after dosing via the indwelling jugular vein catheter into EDTA tubes and stored at −70°C before HPLC-MS analysis of compound 1, M1a-1, and M1a-2 (see above). An aliquot (100 μl) of plasma samples from selected time points (30 min, 2 h, and 6 h) was treated with acetonitrile (200 μl), and samples were centrifuged before transferring the supernatant to another set of clean tubes. The solvents were removed under a stream of nitrogen and the dried residues were reconstituted in 100 μl of the HPLC mobile phase before qualitative LC-MS analysis (see above) was conducted to determine the plasma metabolite profiles.
Pharmacokinetic Analysis.
The pharmacokinetic parameters for individual animals were determined using a noncompartmental analysis module (model 201) of the pharmacokinetic software package WinNonlin (version 5.1; Pharsight, Mountain View, CA). The program applies a model-independent approach, and the standard methods described previously (Gibaldi and Perrier, 1975). The systemic clearance (CLS) was calculated as dose/AUC0–∞ and volume of distribution at steady-state (Vdss) was calculated as mean reaction time (MRT) · CLS, where MRT is AUMC0–∞/AUC0–∞. The area under the plasma concentration versus time curve was calculated using the linear trapezoidal method. Where warranted, the slope of the apparent terminal phase was estimated by log-linear regression using at least three data points and the terminal rate constant (λ) was derived from the slope. AUC0–∞ was estimated as the sum of the AUC0–t (where t is the time of the last measurable concentration) and Ct/λ. The apparent terminal half-life (t1/2) was calculated to be 0.693/λ. For pharmacokinetic calculations, all drug concentration values below the assay limit of quantification were treated as zero.
Statistics.
A least-squares linear regression analysis of the UV peak areas versus concentrations of compounds 1, M1a-1, and M1a-2 were performed using GraFit 5 from Erithacus Software (Horley, Surrey, UK). The concentrations of compounds 1, M1a-1, and M1a-2 in the dosing solutions were determined from the standard curve linear regression equations by inverse prediction using Microsoft Excel 2003. Calculations of means and S.D. were also performed using Microsoft Excel 2003.
Results
Identification and Characterization of Metabolite M1.
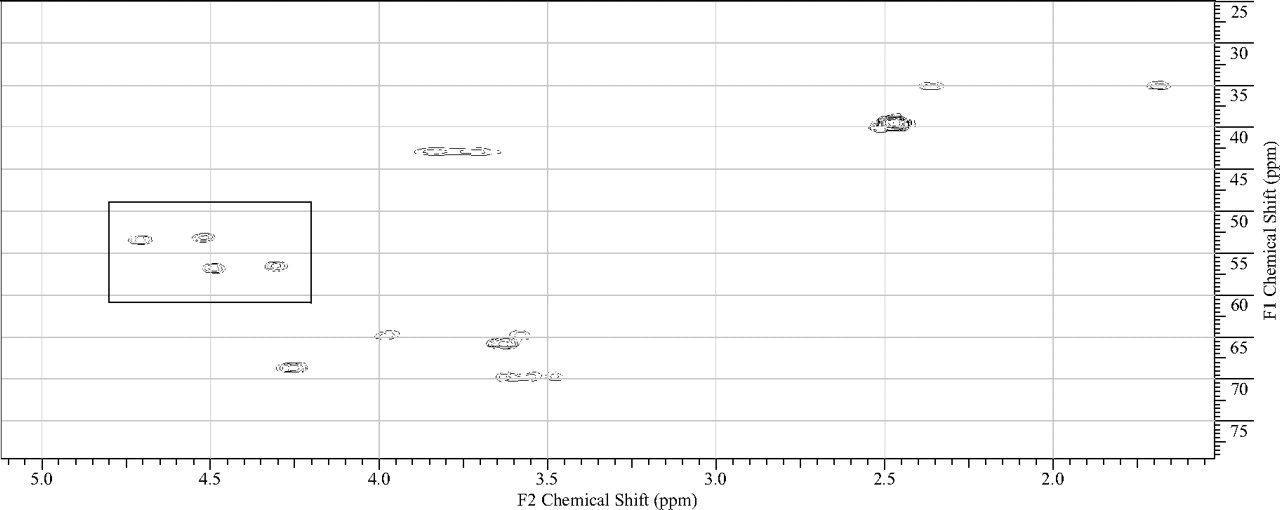
The in vitro metabolism studies conducted with compound 1 showed that it was primarily converted to a single metabolite (M1) in the presence of human and cynomolgus monkey liver microsomes (Fig. 1). Mass spectral analysis of M1 showed that it was a hydroxylated metabolite with [M + H]+ at m/z 505, an increment of 16 atomic mass units from the parent molecular weight. LC-MS analysis conducted using deuterated water showed that there were three exchangeable protons, consistent with the presence of an additional hydroxyl group in metabolite M1 (Fig. 3). Initial NMR analysis of this metabolite showed that hydroxylation had taken place on one of the prochiral carbons forming part of the bridge of the bicyclic ring system. 1H NMR of metabolite M1 (d6-DMSO, 600 MHz): δ 9.22 (s, 1H), 9.20 (s, 1H), 8.37 (d, 2H, J = 7.0 Hz), 8.30 (t, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 6.3 Hz), 7.44 (d, 2H, J = 8.8 Hz), 4.73 (d, 1/2H, J = 7.4 Hz), 4.55 (d, 1/2H, J = 7.3 Hz), 4.51 (d, 1/2H, J = 5.8 Hz), 4.34 (d, 1/2H, J = 5.9 Hz), 4.27 (m, 1H), 3.99 (dd, 1H, J = 16.8, 11.0 Hz), 3.92–3.55 (m, 11H), 2.37 (m, 1H), 1.71 (m, 1H). The hydroxylation of compound 1 produced a chiral center, hence leading to the possibility of enantiomers. The enantiomeric composition of the isolated metabolite M1 could not be determined by NMR analyses. Subsequently, the hydroxylation was shown to have occurred in a very stereospecific manner (see structures in Fig. 2). The stereochemistry of the hydroxylated metabolite was confirmed by chemical synthesis and subsequent chiral separation of resulting enantiomers (Chen et al., 2010). The partial 1H NMR spectra of M1 and two synthetically prepared racemic isomers, M1a (endo) and M1b (exo), are shown in Fig. 4. As shown, the 1H NMR spectrum of the isolated M1 matched that of the racemic endo-isomer, M1a (Fig. 4). The 1H NMR spectrum of the exo-isomer (M1b) was found to be distinctly different from that of the metabolite M1. For the rest of the NMR data for the endo- and exo-isomers of M1, please refer to Chen et al. (2010). Furthermore, it was found that M1 and M1b had different HPLC retention times (although same MS/MS spectra) when analyzed by LC-MS (data not shown). However, it was found that M1 and M1a showed identical retention times on HPLC as well as displaying an identical MS/MS fragmentation pattern (Fig. 3). However, M1a was racemic in nature, and it was important to demonstrate whether the isolated metabolite was racemic or present as a pure enantiomer. Hence, additional studies were conducted to separate the two enantiomers of M1a into M1a-1 and M1a-2 by chiral HPLC (Chen et al., 2010). It was demonstrated clearly by Chen et al. that the biologically produced metabolite M1 corresponded to M1a-1; hence, the formation of metabolite M1 by human liver microsomes seems to have taken place in a very stereospecific manner. Furthermore, each of the enantiomers, M1a-1 and M1a-2, displayed two sets of signals for the bridgehead protons believed to be due to the chair-boat conformations of the bicyclic ring system (Fig. 5). The HSQC data show that the methylene carbon of the bridgehead had two chemical shifts (δ 53.0 and 57.5), and each was correlated to two sets of proton signals.

High-resolution MS/MS spectra of M1 and the endo-isomer (M1a) and exo-isomer (M1b).
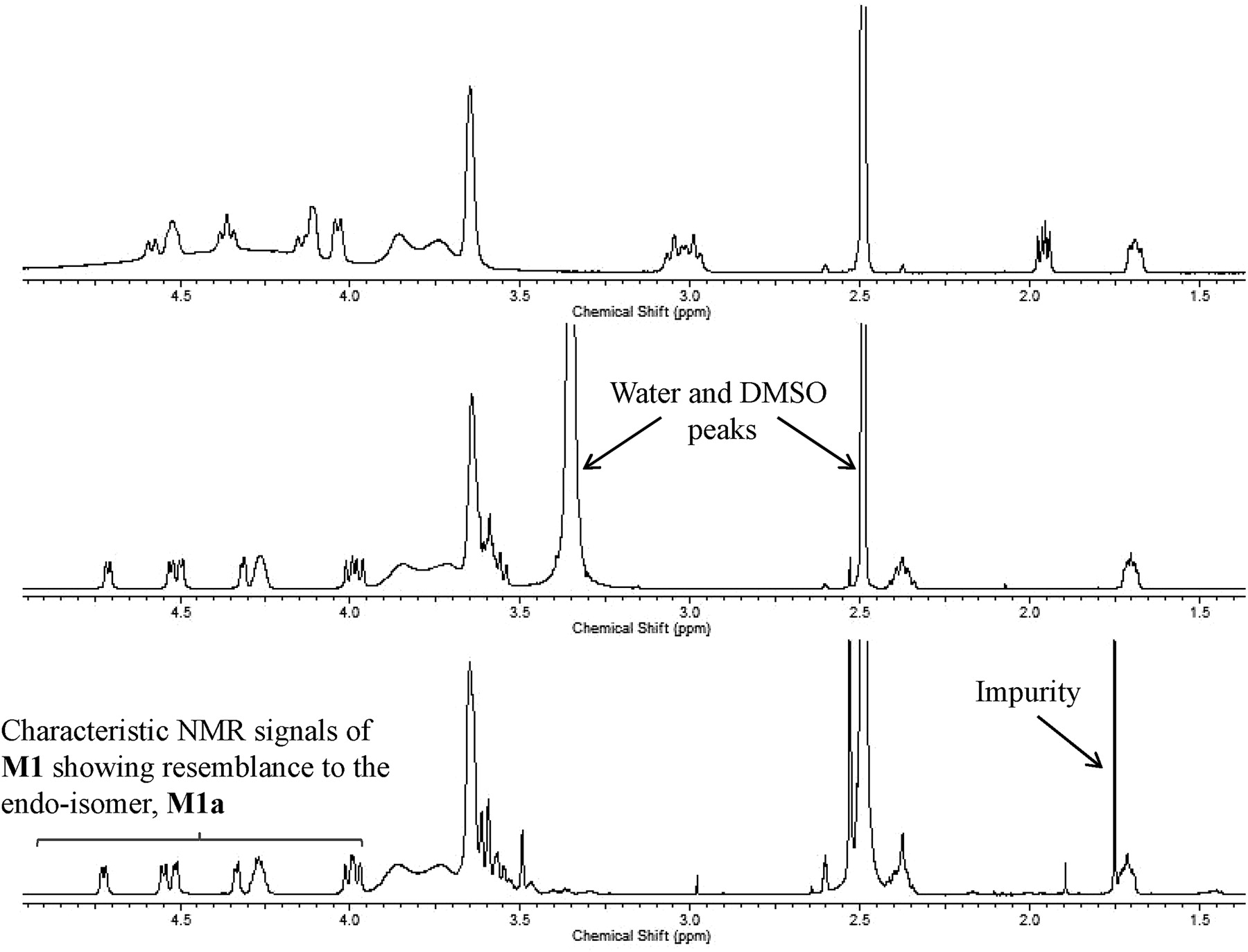
Partial 1H NMR spectra of the aliphatic region of M1 (bottom) and synthetic standards of the endo-isomer M1a (middle), and exo-isomer M1b (top).
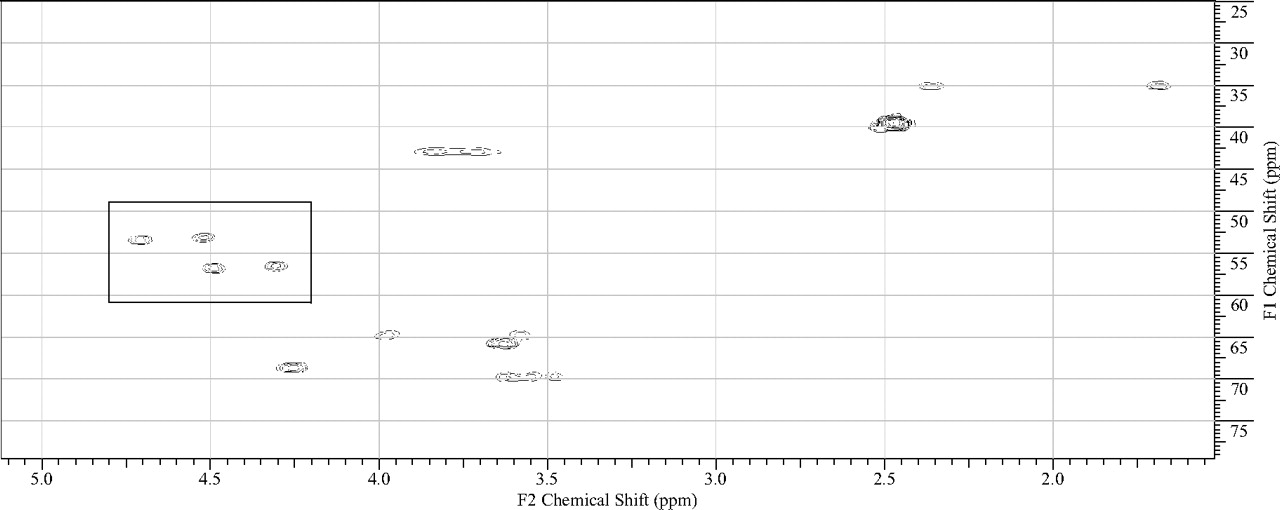
1H-13C HSQC spectrum of M1 showing two different conformations for the bridgehead protons (signals shown within the rectangle) of the bicyclic ring. The same pattern was observed with individual enantiomers M1a-1 and M1a-2 as well as with the racemic mixture, M1a. For the corresponding proton NMR signals, please see Fig. 3.
In Vitro Metabolism of M1a-1 and M1a-2.
Further in vitro metabolism studies were conducted with synthetic standards of enantiomers M1a-1 and M1a-2. LC-MS analysis of microsomal extracts showed that M1a-1 and M1a-2 were both relatively stable in rat liver microsomes, producing only a single hydroxylated metabolite (Fig. 6A). However, M1a-2 was found to be extensively metabolized by monkey and human liver microsomes to a single hydroxylated metabolite (complete structure not determined). In contrast, M1a-1 was found to be relatively stable in the presence of monkey and human liver microsomes (Fig. 6B).

A, LC-MS total ion chromatograms showing that both enantiomers, M1a-1 (bottom) and M1a-2 (top), were metabolized in a similar manner by rat liver microsomes. B, LC/MS total ion chromatograms showing in vitro metabolite profiles of enantiomers, M1a-1 (bottom) and M1a-2 (top), in the presence of human liver microsomes. M1a-2 was highly unstable compared with M1a-1. Similar results were obtained with cynomolgus monkey liver microsomes (data not shown).
Quantitation of Biologically Generated Metabolite M1.
Metabolite M1 was produced in sufficient quantities by human liver microsomes to be characterized and quantitated by NMR analysis. The quantitation of M1 was conducted using the method described previously (Espina et al., 2009). The concentration of M1 was determined to be 1.72 mM (Table 1). A QC sample (another synthetic metabolite standard of compound 1) was used during the NMR analysis to ensure that a reliable concentration of M1 was obtained. Three concentrations of the QC sample (0.2, 2.0, and 3.5 mM) were evaluated by quantitative NMR against gravimetrically determined values. Percent differences between NMR and gravimetrically determined concentrations were found to be less than 15% at all three concentrations.
Pharmacological Activities of the Isolated Metabolite M1 and Chemically Synthesized Metabolites M1a-1 and M1a-2.
For M1, the isolated material showed low nanomolar binding and affinity to the receptor, which was equivalent to that of the parent compound. This initial evaluation using the isolated material was later confirmed by testing the corresponding correct enantiomer, which had in vitro binding similar to that obtained previously using the isolated material (Table 2). Of interest, the epimeric form (M1a-2) of this metabolite was approximately 2 times less active than M1a-1 in three of four pharmacological assays (Table 2). However, it appears that an introduction of a hydroxyl group did not alter the pharmacological activity significantly compared with that for the parent compound.
In Vivo Pharmacokinetics of Compound 1, M1a-1, and M1a-2.
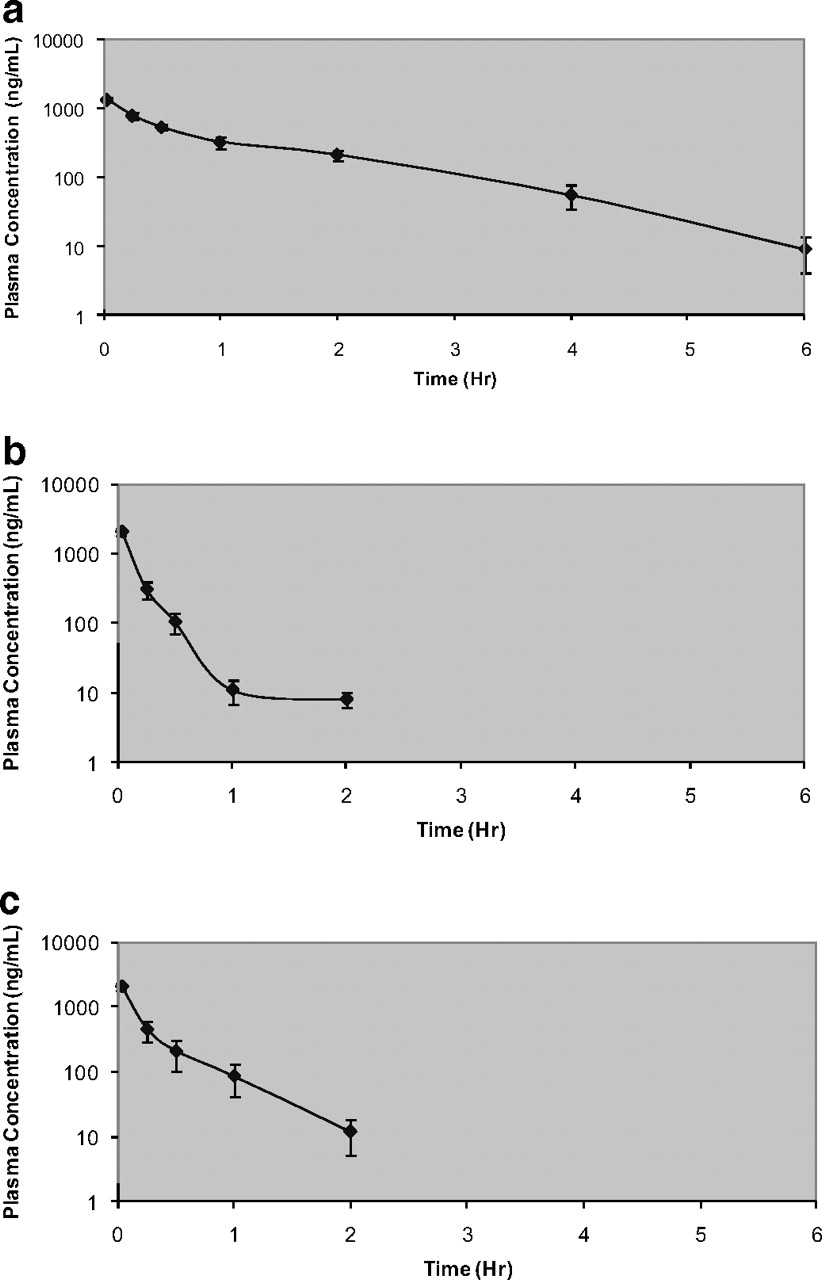
Figure 7, a to c, illustrates semilogarithmic plasma concentration-time profiles for compounds 1, M1a-1, and M1a-2 after 2 mg/kg intravenous bolus doses. The concentration-time profiles were best described by one-compartment models with first-order elimination rate constants. Pharmacokinetic parameters were characterized by high clearance compared with liver blood flow, a moderate volume of distribution compared with plasma volume, and short elimination half-lives (Table 3). Both metabolites had similar clearance and volume of distribution. Oral administration of the compounds (10 mg/kg) produced nondetectable levels of the compound, and hence the data are not shown in this report.

Plasma concentrations of compound 1 (a), M1a-1 (b), and M1a-2 (c) after a single 2 mg/kg intravenous dose in male Sprague-Dawley rats. Compounds M1a-1 and M1a-2 were below the limit of quantitation 2 h postdose.
Pharmacokinetic parameters of compound 1, M1a-1, and M1a-2 in Sprague-Dawley rats after a single 2 mg/kg intravenous dose
Discussion
NMR has traditionally been used to elucidate structures of small and large molecules (Prestegard, 2002; Mutlib and Shockcor, 2003). NMR, especially in combination with mass spectrometry and other hyphenated techniques, is customarily used to fully elucidate structures of previously uncharacterized metabolites (Mutlib and Shockcor, 2003). Although this qualitative aspect of NMR has played a significant role in drug metabolism studies, the quantitative capabilities of NMR in metabolism studies have not been fully realized until recently. With recent improvements in NMR technology, NMR is now considered to be an important quantitative tool despite its lower sensitivity compared with those of the more widely used mass spectrometers. In response to the demands from regulatory agencies to better define the exposures of metabolites in humans and in preclinical species (U.S. Food and Drug Administration, 2008), strategies based on the application of NMR as a quantitative tool for metabolite quantitation have been proposed (Dear et al., 2008; Espina et al., 2009). NMR was used in the past as an analytical tool to determine concentrations of synthetic and biosynthetic products (Warren et al., 1976; Vinson and Kozak, 1978; Werner et al., 1997; Groen et al., 1998; Silvestre et al., 2001; Holzgrabe et al., 2005; Pauli et al., 2005, 2007, 2008; Rizzo and Pinciroli, 2005; Diehl et al., 2007; Shao et al., 2007), metabolites, catabolites, and endogenous compounds in biological fluids (Malet-Martino et al., 1986; Monté et al., 1994; Desmoulin et al., 2002; Orhan et al., 2004; Skordi et al., 2004; Moazzami et al., 2007), or impurities present in products (Hays, 2005; Malz and Jancke, 2006). The theoretical aspects and validation of quantitative NMR have been discussed and reviewed in the literature (Mackenzie, 1984; Evilia, 2001; Burton et al., 2005; Malz and Jancke, 2005).
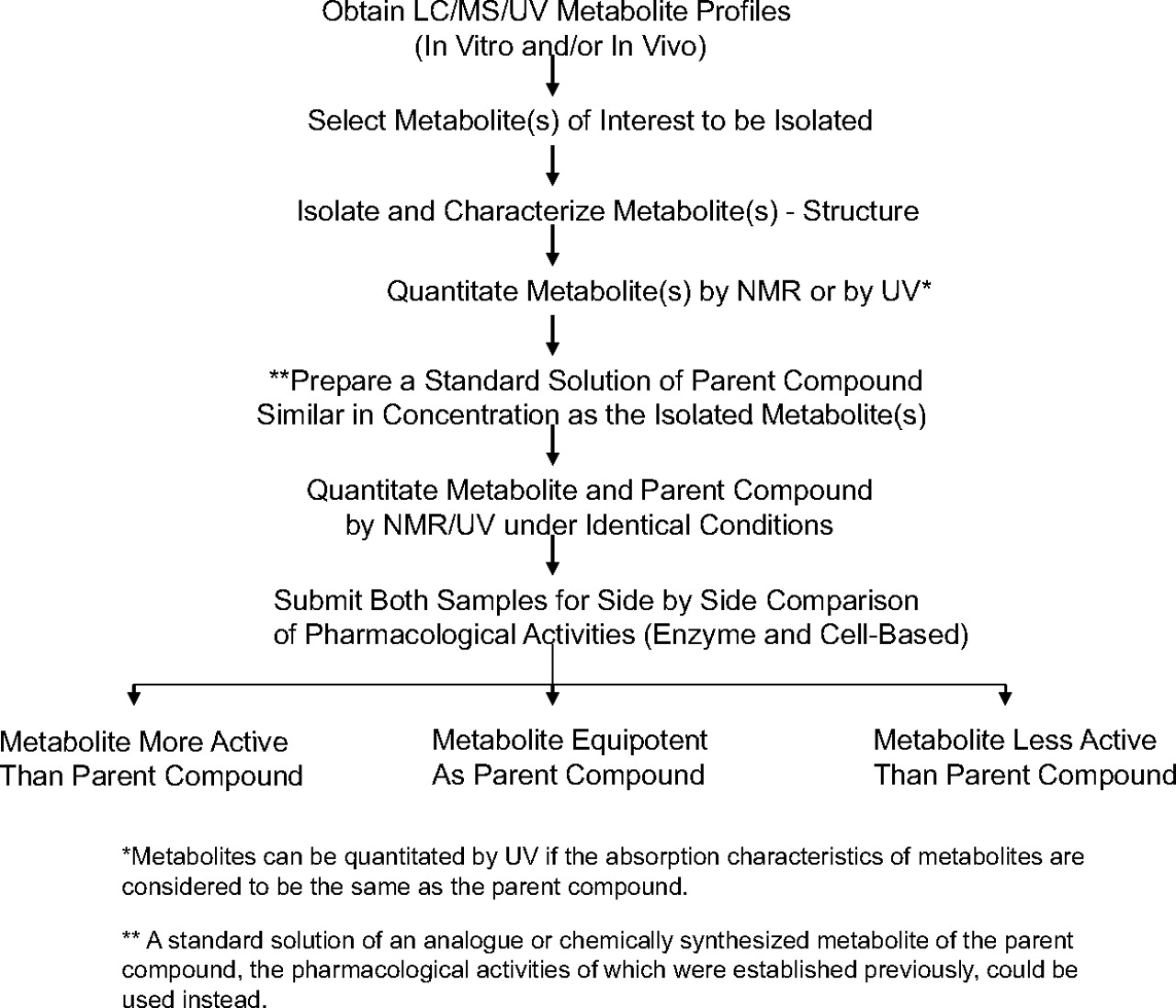
We have demonstrated that NMR could be used to quantitate metabolites isolated from biological sources (Espina et al., 2009). In that study, it was shown that the compounds from which the metabolites are derived could be used to create calibration curves for quantitative analysis. This approach was successfully used to obtain exposure values of metabolites present in plasma of preclinical species, hence circumventing the need for synthetic standards of metabolites or radiolabeled studies. We recently showed the value of conducting quantitative NMR studies in addressing metabolites in safety testing (Vishwanathan et al., 2009). The application of quantitative 19F NMR in obtaining the mass balance of fluorinated compounds was demonstrated previously (Scarfe et al., 1998, 1999; Lenz et al., 2002; Duckett et al., 2006). The ability to quantitate microgram quantities of metabolites led us to further investigate the possibility of using quantitative NMR in accelerating the pharmacological evaluation of metabolites earlier in drug discovery. In the past, pharmacological testing of metabolites was conducted primarily with synthesized metabolite standards. However, frequently, the synthesis of metabolites is protracted, taking several weeks to months before the authentic standards are available for further evaluation. Knowledge of the pharmacological activities of major metabolites earlier can lead to the design of appropriate studies to better understand the implications of such metabolites, especially if they are postulated to have biological activity similar to that of the parent compounds. Once it is known that a metabolite possesses pharmacological activity similar to that of the parent compound (or in some cases more potent), its chemical synthesis can be accelerated to confirm its pharmacological activity. Furthermore, because pharmacologically active metabolites need to be quantitated in clinical samples, the availability of synthetic standards of metabolites in a timely manner can facilitate the assay of this metabolite present in clinical samples. An initial pharmacological evaluation of biologically generated metabolites can be used in the decision-making steps whether chemistry resources are needed for its synthesis. Synthesis of pharmacologically inactive metabolites can be either delayed or not conducted, depending on the fate of the compound as it progresses into development. The other advantage of knowing the pharmacological activity of a metabolite earlier is that it allows one to file a patent on it, hence protecting the intellectual property of a company. Pharmacologically active metabolites can contribute significantly to the overall therapeutic or adverse effects of drugs. Many active metabolites, such as atorvastatin (Lipitor) (Lennernäs, 2003), simvastatin (Zocor) (Williams and Feely, 2002), fluoxetine (Prozac) (Cheer and Goa, 2001), fexofenadine (Allegra) (Meeves and Appajosyula, 2003), and cetirizine (Zyrtec) (Golightly and Greos, 2005) have been developed as drugs in their own right. In an attempt to demonstrate the value of early pharmacological evaluation of biologically derived metabolites, we conducted a series of studies based on initial results with compound 1 and its synthesized metabolite standards. The purpose of these was to demonstrate some of the outcomes and impact of conducting early metabolite identification and quantitation in discovery. The cascade of studies that may occur as a result of confirmatory metabolite identification and quantitation are described in this article (Scheme 1).

Potential studies and outcomes as a result of evaluating pharmacological activities of metabolites earlier in the drug discovery process. SAR, structure-activity relationship; PK, pharmacokinetic.
Compound 1 was found to be metabolized to a major single hydroxylated metabolite by human liver microsomes. As shown in Fig. 1, the hydroxylation took place on a prochiral center, leading to the possibility of several isomers. Although the position of hydroxylation was easily determined by initial NMR analysis of the isolated metabolite, the exact stereochemistry could only be resolved by conducting synthesis and chiral HPLC separation of enantiomers (Chen et al., 2010). After the identity of the metabolite was unequivocally confirmed by HPLC and mass spectral comparisons with synthetic standards, additional studies such as pharmacological and pharmacokinetic evaluations were conducted. It is important to note that it was close to 12 months (from the time the position of hydroxylation on M1 was determined) before synthetic standards of this metabolite were available for pharmacological evaluations. In contrast, it took 3 to 4 weeks from the time the major hydroxylated metabolite was initially identified in human liver microsomes before pharmacological evaluation of the isolated metabolite was conducted. The protracted time required to obtain the synthetic standard of this metabolite was due to a number of reasons including the complex synthesis needed to achieve the desired stereospecificity. Although the initial pharmacological tests on the isolated metabolite were considered to be reliable and accurate, it was important to demonstrate (as part of the verification process) that the synthetic standard produced results that were similar or identical to those for the isolated metabolite in pharmacological evaluations (Table 2). Indeed, the results were very similar and hence confirmed the value of conducting quantitative NMR on isolated metabolites so that pharmacological assessments could be made earlier with such isolated metabolites rather than waiting for synthetic standards of metabolites. Of interest, the introduction of a hydroxyl group on the bridged morpholine ring system did not diminish the pharmacological activity of the metabolite M1. This information was useful to chemists because it led to the design of other molecules with a modified bicyclic ring system (Chen et al., 2010). Fluorinated analogs were also considered so that the formation of the hydroxylated metabolite could be mitigated. By evaluating the pharmacological activities of metabolites, one may also gain an insight into structure-activity relationships; hence potentially influencing the design of newer molecules (Scheme 1). For example, in the case of compound 1, fluorine and methyl groups were substituted in the metabolic soft spot, realizing that introduction of a small group at that position was not going to compromise the pharmacological activity. Further studies with these analogs are in progress. Often the chemical synthesis of a metabolite(s) is accelerated once it is realized that it possesses pharmacological activity (Scheme 1). This in turn allows additional testing of the metabolite in a pharmacology model. Other studies such as in vitro metabolic disposition or in vivo pharmacokinetic evaluation of these synthetic metabolite standards could then easily be conducted. The information derived from all of these studies can lead to better understanding of the disposition of the parent compound as well its pharmacologically active metabolite in preclinical species. In some cases, the properties of metabolite (or other synthesized) analogs may be more favorable than those of the parent compound; hence potentially creating a new drug candidate. With compound 1, it was found that the major metabolite M1 was produced in a very stereospecific manner, existing as the enantiomer M1a-1. Furthermore, it was shown that this enantiomer was more metabolically stable than M1a-2, which was shown not to be produced as a metabolite. The racemic mixture (M1a) also displayed liver microsomal instability because of the rapid metabolism of the enantiomer M1a-2. If this hydroxylated metabolite was further pursued as a potential drug candidate, it would be desirable to synthesize the more stable enantiomer, M1a-1. The significant difference in the metabolic disposition of the two enantiomers is consistent with what has been reported before for other racemic compounds (Mutlib and Nelson, 1990 and references cited therein).
Pharmacokinetic studies showed that metabolite M1 (M1a-1 enantiomer) had higher clearance and lower exposure values than the parent compound 1. The overall pharmacokinetic behavior of the two enantiomers, M1a-1 and M1a-2, were similar. This observation was consistent with in vitro metabolism data, which showed similar metabolic stability in the presence of rat liver microsomes. Furthermore, the in vitro metabolism data suggested that these hydroxylated metabolites (M1a-1 and M1a-2) were not predisposed to glucuronidation as evidenced by lack of glucuronide conjugates in the metabolite profile (Fig. 6A). Furthermore, the LC/MS analysis of plasma extracts obtained from the pharmacokinetic studies showed an absence of any glucuronide conjugates, consistent with in vitro metabolism data (data not shown). It is postulated that the high clearance of M1a-1 and M1a-2 could be due to their rapid elimination in bile, although further studies will be needed to confirm this. The high clearance of this metabolite (M1a-1) observed in rats led to termination of this compound as a viable candidate for further development.
A strategy is proposed to ensure that reliable pharmacology results are obtained from metabolites that are isolated from biological sources (Scheme 2). Once a metabolite of interest is selected, it is proposed that a scale-up and subsequent isolation and characterization take place to confirm the structure of the metabolite. The NMR analysis of the isolated metabolite is used to confirm the identity (and also purity) of the isolated metabolite. Subsequently, the metabolite can be quantitated by HPLC-UV if the absorption characteristics of the metabolite and the parent compound are considered to be similar. In general, if the site of metabolism is remote from the UV chromophore, the absorption characteristics are relatively identical for the parent compound and its metabolite(s). If the metabolite UV spectrum is not the same as that of the parent compound, 1H NMR can be used to generate the calibration curve for quantitation purposes (Espina et al., 2009). The parent compound, in this case compound 1, can be used to generate the standard calibration curve, which could be used to quantitate the isolated metabolite (Espina et al., 2009). A standard solution of the parent compound similar in concentration to that of isolated metabolite is made, and both preparations are subjected to quantitative analysis by NMR. Once the concentrations are verified, both samples are submitted for side-by-side comparison of pharmacological activities (Scheme 2). The preparation of a separate standard solution of the parent compound (such as compound 1) acts as a quality control for both NMR analysis and also for pharmacological evaluation. As an alternative, to ensure the integrity of the results, an analog of the parent compound or synthetically available metabolite standard could be weighed and its concentration verified by NMR so that pharmacological evaluations could be performed (Table 1). In the case of compound 1, a synthetic standard of metabolite that was used as the QC for quantitative NMR was found to be inactive (data not shown).

Workflow demonstrating the application of quantitative NMR in evaluating pharmacological activities of isolated metabolites for comparison with parent compound.
The value of performing quantitative NMR on isolated metabolites for the purpose of further pharmacological tests was demonstrated. A better understanding of the disposition of compound 1 and its metabolite M1 was achieved once it was demonstrated that M1 was pharmacologically active. Because sufficient quantities of M1 were readily available for quantitation as well as for method development, the synthesis and identification of the correct enantiomer was greatly facilitated. Subsequent pharmacokinetic evaluations of the pharmacologically active chemically synthesized metabolite showed that it had the propensity to be rapidly cleared in vivo, making it an unlikely candidate for further development. Overall, as illustrated with this example, design of experiments and critical resource-sparing decisions could be facilitated if pharmacological activities of metabolites are known much earlier in the drug discovery and development process.
Authorship Contributions
Participated in research design: Mutlib, Espina, Dehnhardt, Venkatesan, and Chaudhary.
Conducted experiments: Espina, Vishwanathan, Babalola, Chen, and Chaudhary.
Contributed new reagents or analytic tools: Espina.
Performed data analysis: Mutlib, Espina, Venkatesan, and Chaudhary.
Wrote or contributed to the writing of the manuscript: Mutlib, Espina, Chen, Dehnhardt, Venkatesan, and Chaudhary.
Other: Mansour, Talaat, and Scatina acquired funding for the research and reviewed manuscript.
Footnotes
- Received February 1, 2010.
- Accepted October 15, 2010.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032490.
ABBREVIATIONS:
- LC
- liquid chromatography
- MS
- mass spectrometry
- HPLC
- high-performance liquid chromatography
- MS/MS
- tandem mass spectrometry
- QC
- quality control
- HSQC
- heteronuclear single quantum correlation
- AUC
- area under the plasma concentration versus time curve
- DMSO
- dimethyl sulfoxide.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}