


Abstract
In a previous study, we used human liver microsomes for the first time to study cytochrome P450 (P450)-mediated oxidation of the flavonoid galangin. The combination of CYP1A2 and CYP2C9 produced aVmax/Km value of 13.6 ± 1.1 μl/min/mg of protein. In the present extended study, we determined glucuronidation rates for galangin with the same microsomes. Two major and one minor glucuronide were identified by liquid chromatography/mass spectrometry. TheVmax/Km values for the two major glucuronides conjugated in the 7- and 3-positions were 155 ± 30 and 427 ± 26 μl/min/mg of protein, thus, exceeding that of oxidation by 11 and 31 times, respectively. This highly efficient glucuronidation appeared to be catalyzed mainly by the UDP-glucuronosyltransferase (UGT)1A9 isoform but also by UGT1A1 and UGT2B15. Sulfation of galangin by the human liver cytosol, mediated mainly but not exclusively by sulfotransferase (SULT) 1A1, also appeared to be efficient. These conclusions were strongly supported by experiments using the S9 fraction of the human liver, in which all three metabolic pathways could be directly compared. When galangin metabolism was examined in fresh plated hepatocytes from six donors, glucuronidation clearly predominated followed by sulfation. Oxidation occurred only to a minor extent in two of the donors. This study for the first time establishes that glucuronidation and sulfation of galangin, and maybe other flavonoids, are more efficient than P450-mediated oxidation, clearly being the metabolic pathways of choice in intact cells and therefore likely also in vivo.
Dietary flavonoids have long been implicated to be preventive in disease processes, including, most prominently, cardiovascular disease and cancer. The bioavailability of the dietary flavonoids, however, is low (Manach et al., 1998; Erlund et al., 2000; Walle et al., 2000b; Graefe et al., 2001), limited both by poor absorption (Walgren et al., 1998) and extensive metabolism (Walle et al., 1999). The metabolism of the flavonoids appears to include oxidative as well as conjugative metabolism, but there is little systematic information on the metabolism of these dietary compounds in humans. As no previous studies had been conducted with human liver microsomes, we recently examined the oxidation of several flavonoids by this preparation (Otake and Walle, 2002). One of these flavonoids, the flavonol galangin, was extensively oxidized in the 4′-position to kaempferol (Fig.1) with an apparentKm value of 9.5 ± 0.4 μM and aVmax/Kmvalue of 13.6 ± 1.1 μl/min/mg of protein. This reaction was catalyzed mainly by cytochrome P450 CYP1A2 but also by CYP2C9.

Structures of galangin and its oxidation product kaempferol.
The primary objective of the present study was to examine the metabolism of galangin by glucuronidation, using the same human liver microsomes as well as recombinant UDP-glucuronosyltransferase (UGT1) isoforms. Also, we determined the role of sulfation, using the human liver cytosol as well as recombinant sulfotransferase (SULT) isoforms. We compared all three pathways (i.e., oxidation, glucuronidation, and sulfation) in the S9 fraction of the human liver. Finally, we determined the relative contribution of each of these pathways to galangin metabolism in fresh plated human hepatocytes from six donors.
Materials and Methods
Chemicals.
Galangin, d-glucose 6-phosphate,d-glucose-6-phosphate dehydrogenase, kaempferol, NADP, uridine 5′-diphosphoglucuronic acid (UDPGA), alamethicin, saccharic acid 1,4-lactone, ethoxyresorufin, resorufin, salicylamide, tetrabutylammonium hydrogen sulfate, and trifluoroacetic acid (spectrophotometric grade) were purchased from Sigma-Aldrich (St. Louis, MO). Ultra pure 3′-phosphoadenosine-5′-phosphosulfate (PAPS) was obtained from S. S. Singer, University of Dayton, Dayton, OH. [35S]PAPS (1–3 Ci/mmol) was obtained from PerkinElmer Life Sciences (Boston, MA). Pooled human liver cytosol, microsomes, and S9 fraction from 10, 11, and 10 donors, respectively, and recombinant human UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15 were purchased from Gentest Corp. (Woburn, MA), and UGT1A3 was purchased from PanVera Corp. (Madison, WI).
Glucuronidation by Human Liver Microsomes, Human Liver S9 Fraction, and Recombinant UGTs.
Galangin (0.1–100 μM) was preincubated with pooled human liver microsomes (3 μg of protein), pooled human liver S9 fraction (3 μg of protein), or recombinant UGTs (5 μg of protein) in 50 mM Tris buffer (pH 7.4) with 10 mM MgCl2 for 5 min at 37°C in a total reaction volume of 100 μl. The reaction was then initiated by addition of 1 mM UDPGA and incubated for 15 to 30 min at 37°C. Controls were incubated in the absence of UDPGA. To terminate the reaction, an equal volume of cold methanol was added, and the samples were centrifuged at 14,000g for 2 min. The supernatant was subjected to HPLC analysis. In some experiments, incubations were done in the presence of 50 or 150 μg of alamethicin/mg of microsomal protein and 5 mM saccharolactone, as previously described (Fisher et al., 2000). Kinetic parameters were obtained by fitting the data to the Michaelis-Menten equation using the Solver function in Excel (Microsoft, Redmond, WA).
Sulfate Conjugation by Human Liver Cytosol, Human Liver S9 Fraction, and Recombinant SULTs.
Galangin (0.01–5 μM) was incubated with pooled human liver cytosol or pooled human liver S9 fraction (50 μg of protein each) at 37°C for 30 min in 33 mM Tris buffer (pH 7.4) containing 0.0625% BSA, 8 mM dithiothreitol, and 1 μM [35S]PAPS in a total reaction volume of 100 μl. Sulfate conjugates were extracted as ion pairs with tetrabutylammonium hydrogen sulfate and ethyl acetate (Varin et al., 1987), and the radioactivity in the extract was measured by liquid scintillation spectrometry. Recombinant human P-form phenol sulfotransferase (SULT1A1, P-PST) was prepared as previously described (Lewis et al., 1996). Recombinant human M-form phenol sulfotransferase (SULT1A3, M-PST) (Ganguly et al., 1995), human dehydroepiandrosterone sulfotransferase (SULT2A1, DHEA-ST) (Comer et al., 1993), and human estrogen sulfotransferase (SULT1E1, EST) (Falany et al., 1995) were purified from pkk233–2 constructs obtained from Dr. C. N. Falany and expressed in Escherichia coli. Recombinant SULTs were incubated with galangin under identical conditions, using 0.65 μg of SULT1A1 (total protein), 2.6 μg of SULT1A3, 2.2 μg of SULT2A1, or 0.63 μg of SULT1E1 in a total reaction volume of 100 μl. In separate experiments, the ethyl acetate extract above was taken to dryness under nitrogen gas and reconstituted in mobile phase, which was then subjected to HPLC analysis (see below) for further confirmation of intact sulfate conjugate.
Incubation of Galangin with Fresh Human Hepatocytes.
Fresh human hepatocytes were purchased from In Vitro Technologies Inc. (Baltimore, MD). These hepatocytes, as well as human liver microsomes, were prepared from nontransplantable livers of clinically dead donors. Of six donors (two males and four females), four died of cerebrovascular accident, one of head trauma, and one of stroke. The cells were plated in 12 wells at a density of 700,000 cells per 1-cm2 well. After attachment, the cells were shipped overnight at room temperature with the wells filled with culture medium. Upon arrival of cells within 36 to 48 h after plating, the plates were checked for loose cells, and the medium was replaced. The plates were put in a humidified 37°C incubator with 5% CO2 for 1 h to recover from shipping. The medium was then replaced with 0.6 ml of the same serum-free medium containing 25 μM galangin. After 1, 3, and 6 h incubation, 100-μl aliquots of the medium were treated with an equal volume of methanol to precipitate protein and centrifuged for 2 min at 14,000g. The supernatant (150 μl) was subjected to HPLC analysis. Galangin metabolites were identified by LC/MS (see below). Quantitation of galangin metabolites was based on peak areas compared with a standard curve obtained for galangin. The cells were digested with 0.5 M NaOH and analyzed for protein content (Lowry et al., 1951).
HPLC Analysis.
Galangin samples were analyzed by reverse-phase HPLC using a Millennium HPLC system (Waters Corp., Milford, MA) with a photodiode array detector (Waters model 996) and a Symmetry C18column (3.9 × 150 mm; Waters Corp.) with a μBondapak C18 GuardPak precolumn insert (Waters Corp.). Galangin and its metabolites were eluted in a mobile phase of 60% methanol and 0.3% trifluoroacetic acid at a flow rate of 0.9 ml/min and were detected at 370 nm. This wavelength gave high specificity for galangin and its metabolites compared with endogenous compounds. However, as the pure galangin metabolites were not available, we had to assume that their molar extinction coefficients were the same as for galangin. This likely led to an underestimation, in particular of glucuronic acid conjugate peak 1, for which the UV absorption decreased in comparison with that of galangin.
LC/MS Analysis.
LC/MS analysis was performed using an HP1100 LC binary pump chromatography system (Hewlett Packard, Palo Alto, CA) coupled with a triple quadrupole mass spectrometer API300 (Applied Biosystems, Foster City, CA). The mass spectrometer was interfaced with a turbo ionspray ionization source and operated in a positive ionization mode with the ionization potential of 4 kV and drying temperature at 300°C. Galangin and metabolites were separated on a YMC ODS-AQ column (50 × 2.0 mm i.d., Waters Corp.) using a mobile phase of a linear gradient of 5 to 90% solvent B (0–10 min) at a flow rate of 0.2 ml/min. Solvent A was 0.5% acetic acid in water and solvent B was 0.5% acetic acid in acetonitrile. The tandem MS experiments were operated using nitrogen as collision gas with collision energy of 85 eV. The constant neutral loss tandem MS experiment with a mass offset of 176 atomic mass units was performed to detect glucuronic acid conjugates. The daughter ion scans at m/z 447 (galangin glucuronide) and at m/z 271 (galangin) were performed to confirm galangin conjugates.
CYP1A2 Fluorometric Assay.
Hepatocytes were incubated with 0.6 μM ethoxyresorufin for 30 min in the presence of 1.5 mM salicylamide (Ciolino et al., 1998). The formation of resorufin was measured fluorometrically in the medium with excitation/emission at 530:590 nm.
Results
Glucuronic Acid Conjugation of Galangin by Human Liver Microsomes and Recombinant UGTs.
When 10 μM galangin was incubated with human liver microsomes (pooled from 11 donors) in the presence of the cofactor UDPGA and subjected to HPLC, three peaks (1–3) were observed, which were not present in the absence of UDPGA (Fig. 2, top). These peaks were identified by LC/MS as glucuronic acid conjugates of galangin. They all had molecular ions (M + H)+ ofm/z 447, which underwent collision-induced fragmentation by losing the glucuronic acid moiety of 176 atomic mass units to produce ions of m/z 271, all highly characteristic of glucuronic acid conjugates. In addition, the peaks were also effectively hydrolyzed by β-glucuronidase to yield galangin. Kinetics of the formation of the major peaks 1 and 2 by the liver microsomes are shown in Fig. 3A (mean ± S.E. of three experiments). Kinetic parameters obtained by fitting the data to the Michaelis-Menten equation, using the Solver function in Excel, are shown in Table 1. Although the Km value for peak 1 was considerably lower than for peak 2, itsVmax value was also much lower, yielding apparent efficiencies (i.e.,Vmax/Kmvalues) that were similar and very high, 427 and 155 μl/min/mg of protein, respectively.

HPLC/UV analysis of 1-h incubates of human liver microsomes (HLM) (10 μg of protein) and recombinant UGT isoforms (20 μg of protein) with 10 μM galangin and 1 mM UDPGA.
Samples were analyzed directly after precipitation of proteins with methanol. Peaks 1, 2, and 3 were not present in the absence of UDPGA. AU, absorbance units.
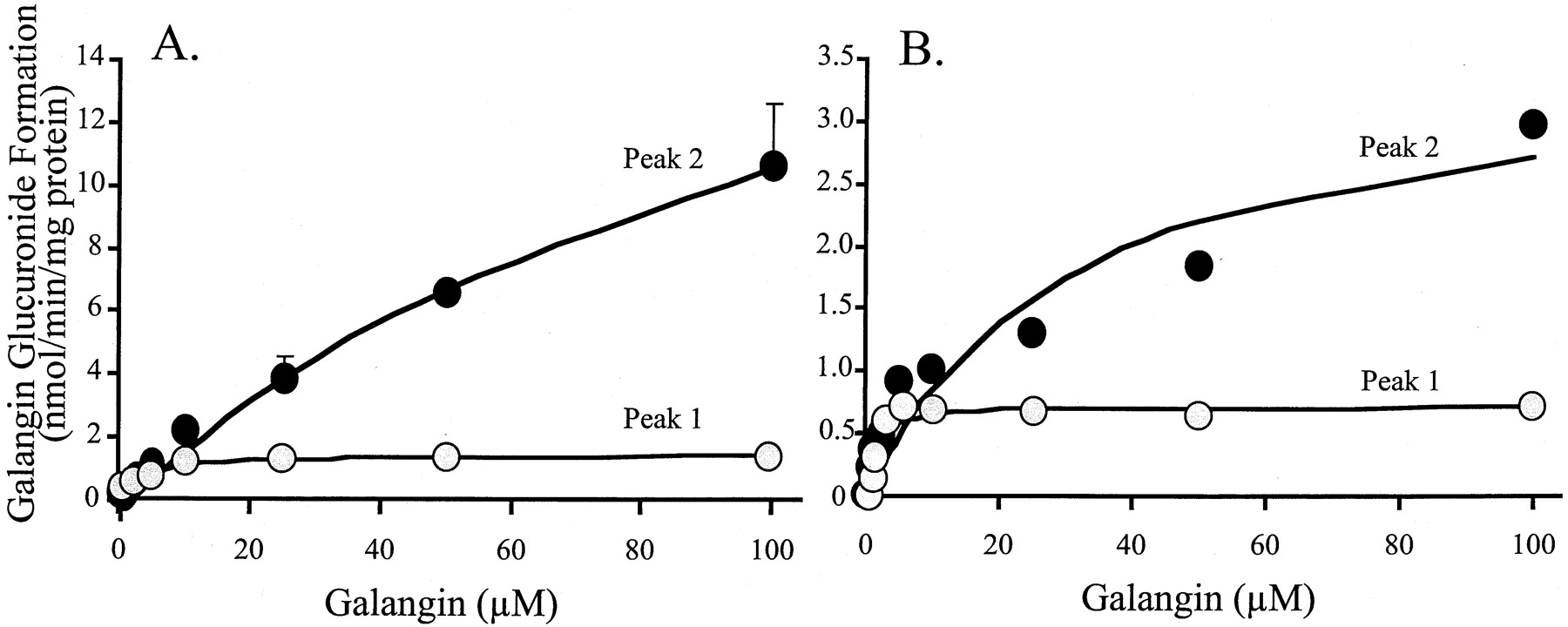
Velocity of galangin glucuronidation versus substrate concentration for (A) human liver microsomes and (B) recombinant UGT1A9.
Data are mean values ± S.E. for three determinations with the human liver microsomes (pooled from 11 donors) and mean values for two determinations with UGT1A9. Peaks 1 and 2 refer to Fig. 2.
Apparent enzyme kinetic parameters for glucuronidation of galangin by HLM and recombinant UGT1A1, 1A9, and 2B15
To determine which UGT isoform(s) may be responsible for this high glucuronidation activity, we examined first the three isoforms that previously had been shown to use another similar flavonoid (i.e., chrysin) as substrate, namely UGT1A1, UGT1A6, and UGT1A9 (Walle et al., 2000a). As can be seen in Fig. 2, recombinant UGT1A1 produced all three glucuronides, although in different ratios than the human liver microsomes, and recombinant UGT1A6 produced small amounts of peaks 2 and 3 only. Recombinant UGT1A9, on the other hand, produced peaks 1 and 2 in a ratio similar to that of the human liver microsomes. Based on consumption of the parent compound galangin (Fig. 2), UGT1A9 was by far the most efficient of these isoforms. Of the additional UGT1A isoforms present in the human liver (Tukey and Strassburg, 2001), UGT1A3 produced small amounts of peaks 1 and 2, and UGT1A4 was inactive. In addition, two UGT2B isoforms available to us both showed activity, UGT2B7 forming small amounts of peaks 1 and 2 but UGT2B15 forming substantial amounts of peak 2 only, Fig. 2.
The kinetics of galangin glucuronidation by the most efficient of the isoforms in Fig. 2 (i.e., UGT1A9, mean values of two experiments) are shown in Fig. 3B, demonstrating similar behavior as the glucuronidation by the human liver microsomes in Fig. 3A. Whereas formation of peak 1 follows Michaelis-Menten kinetics, peak 2 shows some deviations. The reason for the latter is not clear. When repeating this experiment with an improved preparation of recombinant UGT1A9 (i.e., a baculovirus preparation), there was a much better fit of the data. Nevertheless, the apparent kinetic constants calculated from these data are shown in Table 1. For comparison, the kinetics of peak 2 formation by UGT1A1 and UGT2B15 are also shown in this table. Whereas UGT1A9 appears to be the main contributor by far to peak 1 formation in the liver, there is a similar contribution by UGT1A1, UGT1A9, and UGT2B15 to peak 2 formation. These conclusions, however, have to be guarded, as the absolute amount of recombinant protein present in each preparation is unknown.
Previous studies of UGT activities have demonstrated that for some substrates and/or UGT preparations, it is important to consider difficulties of the substrate to enter the microsomal lumen as well as competing β-glucuronidase activity (Fisher et al., 2000). When experiments were carried out in the presence of the pore-forming alamethicin and the β-glucuronidase inhibitor saccharolactone, there was only a minimal increase (<25%) of UGT activities in our preparations.
UV spectroscopy has been shown to be a useful and highly sensitive diagnostic tool to establish the site of certain substitutions in flavonoid molecules (Mabry et al., 1970), most recently applied to glucuronic acid conjugates of flavopiridol (Jäger et al., 1998) and chrysin (Galijatovic et al., 1999). Galangin shows two absorption maxima at 253 (band II, 4-phenyl-3-hydroxyl-2,3-double bond) and 359 (band I, 5,7-dihydroxyphenyl) nm. In peak 1, band II remained the same, but the intensity of band I diminished compared with band II to what is typical of a flavone, rather than a flavonol. This is consistent with a glucuronide in the 3-position. In peak 2, there was no change in either band II or I, consistent with a glucuronide in the 7-position. In peak 3, both band II and band I had shifted about 5 nm toward shorter wavelength, consistent with a glucuronide in the 5-position (Mabry et al., 1970).
Sulfate Conjugation of Galangin by Human Liver Cytosol and Recombinant SULT Isoforms.
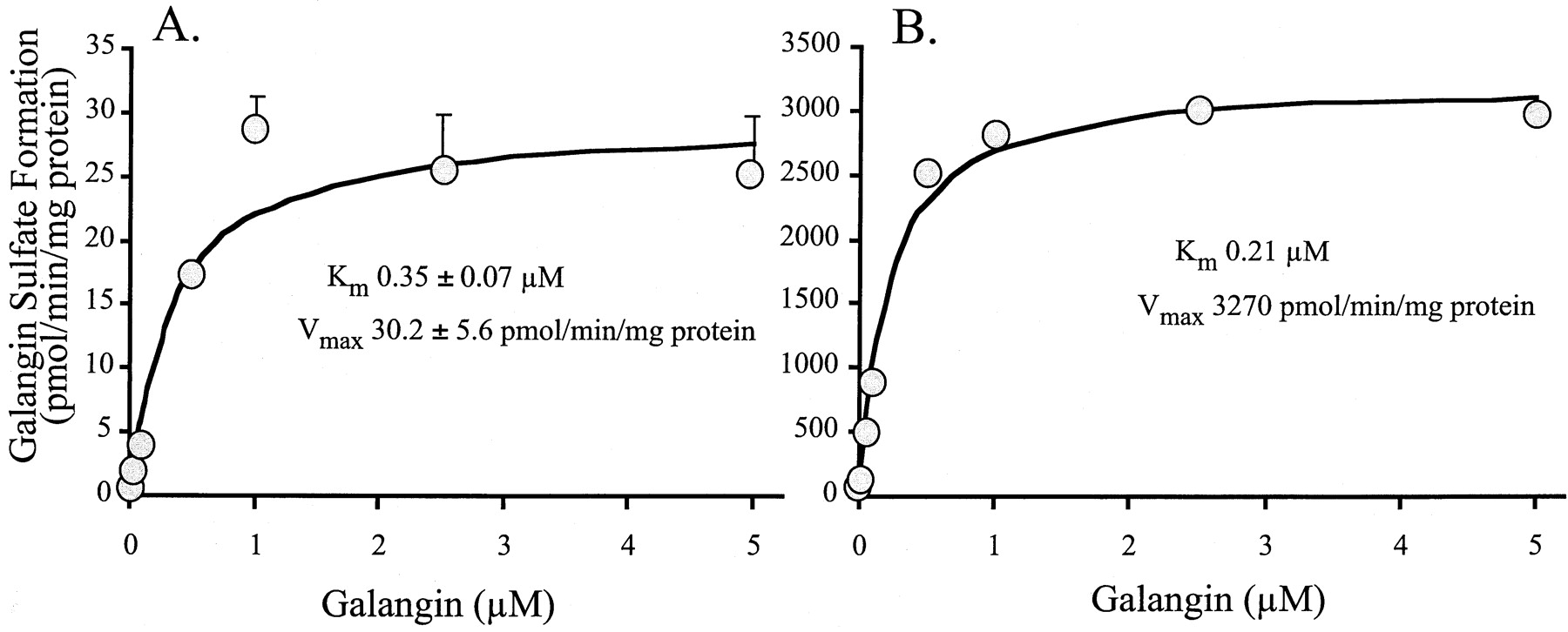
Conjugation with sulfate would be expected to be a metabolic pathway competing with the glucuronidation of galangin. As the SULTs are soluble enzymes, this was examined with the human liver cytosol (pooled from 10 donors) in the presence of 1 μM cofactor PAPS. As seen in Fig. 4B, this resulted in a single peak by HPLC, which was not present in the absence of PAPS (Fig. 4A). Also, when using 35S-labeled PAPS in experiments similar to those described previously (Walle et al., 1993; Galijatovic et al., 1999), the peak in Fig. 4B became radiolabeled. Using radiolabeled PAPS, we next determined the kinetics of galangin sulfation by the method ofVarin et al. (1987), Fig. 5A. A very lowKm value of 0.35 ± 0.07 μM was obtained. The main SULT isoform in the human liver is SULT1A1 (P-PST) (Campbell et al., 1987; Falany, 1997). When sulfation of galangin by recombinant SULT1A1 was determined (Fig. 5B), a similar low apparentKm value of 0.21 μM was obtained as with the human liver cytosol. As several other SULT isoforms may contribute, we also examined recombinant SULT1A3 (M-PST), SULT2A1 (DHEA-ST), and SULT1E1 (EST) for activity with galangin. These results are summarized in Table 2. Thus, SULT1E1 had a rather low Km value, whereas for SULT1A3 this was considerably higher. SULT2A1 had no activity with this substrate. It should be pointed out that, as with recombinant UGT isoforms, the levels of the recombinant SULT isoforms in the individual preparations are not known, except for SULT1A1, which is >90% pure (Lewis et al., 1996). The Vmax andVmax/Kmvalues are therefore not comparable.

HPLC/UV analysis of 30-min incubates of 5 μM galangin with human liver cytosol (pooled from 10 donors) (A) in the absence and (B) in the presence of 1 μM PAPS.
AU, absorbance units.

Velocity of galangin sulfonation versus substrate concentration for (A) human liver cytosol and (B) recombinant SULT1A1 (P-PST).
Data are mean values ± S.E. for four determinations with the human liver cytosol (pooled from 10 donors) and mean values for two determinations with SULT1A1.
Apparent enzyme kinetic parameters for sulfation of galangin by SULT1A1, 1A3, 2A1, and 1E1
Metabolism of Galangin by the S9 Fraction of Human Livers.
To be able to assess the metabolism of galangin by all three metabolic routes (i.e., oxidation, glucuronidation and sulfation) in a single preparation, we used the pooled S9 fraction from 10 donors. The kinetic data derived (n = 3 for all) are summarized in Table3. The oxidation had an apparentKm value of 10.5 μM with aVmax/Kmvalue of 8.0 μl/min/mg of S9 protein. The formation of peak 1 by glucuronidation had a Km value of 2.8 μM, (i.e., similar to what was observed with the microsomes in Table1) and aVmax/Kmvalue of 230 μl/min/mg of S9 protein. The formation of peak 2 by glucuronidation had a lower Km value (i.e., 36.4 μM) than seen with the microsomes. TheVmax/Kmvalue for peak 2 was 133 μl/min/mg of S9 protein. The sulfation of galangin by the S9 fraction was quite similar to that seen with the cytosol in Fig. 5.
Apparent enzyme kinetic parameters for oxidation, glucuronidation and sulfation of galangin by the S9 fractions of human livers
Metabolism of Galangin by Human Hepatocytes.
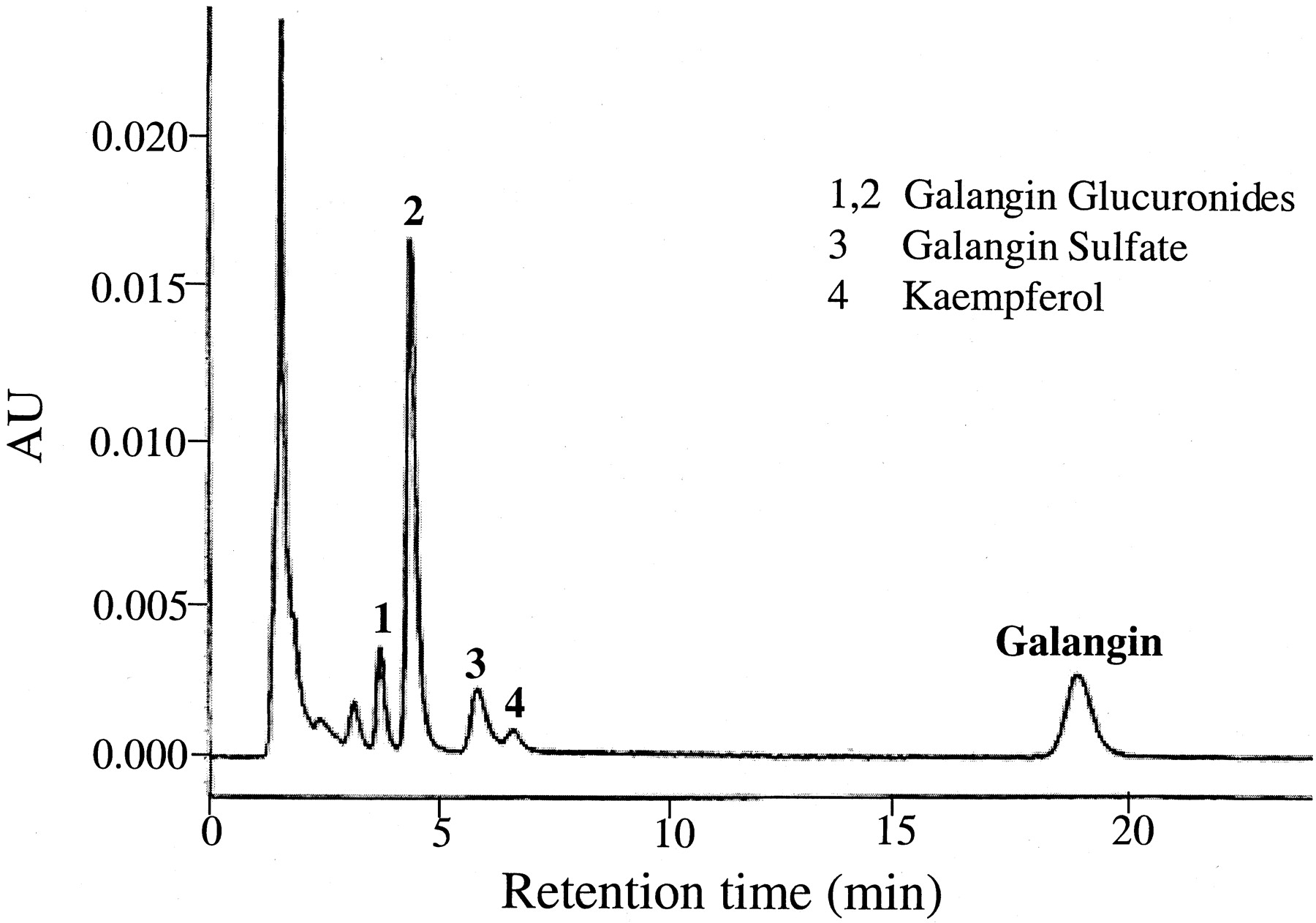
Having determined the qualitative as well as the quantitative metabolic fate of galangin by P450-mediated oxidation (Otake and Walle, 2002) as well as glucuronidation and sulfation, using human liver subcellular and cellular fractions, we tested our observations in plated, fresh human hepatocytes. Figure 6shows an HPLC tracing of galangin and its metabolites formed after a 3-h incubation with human hepatocytes from one donor. The parent compound and four metabolites (1–4) could be detected (i.e., were not present in control incubates without galangin). The major metabolites, peaks 1 and 2, were identical to the glucuronic acid conjugate peaks in Fig. 2, formed by the human liver microsomes, with respect to retention times as well as characteristic UV spectra. Peak 3 was identical to galangin sulfate in Fig. 4, using the same criteria. The smallest peak 4 was difficult to identify, as it appeared to contain two metabolites of galangin. Using UV spectra as criteria, this peak in most donors corresponded to the minor glucuronide (peak 3) in Fig. 2. In two of the donors, this peak appeared to contain kaempferol (i.e., the 4′-hydroxylation product of galangin). This was the only metabolite formed when galangin was incubated with human liver microsomes and an NADPH-generating system (Otake and Walle, 2002). The metabolic data for all six hepatocyte donors studied are summarized in Fig.7. Quantitation of the major metabolites formed (i.e., peaks 1, 2, and 3 in Fig. 6) and galangin were based on UV absorption at 370 nm (see Materials and Methods). Galangin was efficiently metabolized by the hepatocytes with less than 10% galangin remaining at 6 h. Glucuronidation (peaks 1 and 2 in Fig. 6) accounted for more than 70% of the metabolism, with sulfation (peak 3) accounting for about 15%. P450-mediated oxidation (peak 4 in Fig. 6) only occurred to a minor extent in two of the six hepatocyte donors (e.g., in the donor in Fig. 6). Overall, there was about a 5- to 8-fold interindividual variability in galangin metabolism in this group of six subjects. This variability could not be related to the limited phenotypic differences available for the individual tissue donors, such as smoking, alcohol intake, or therapeutic drugs.
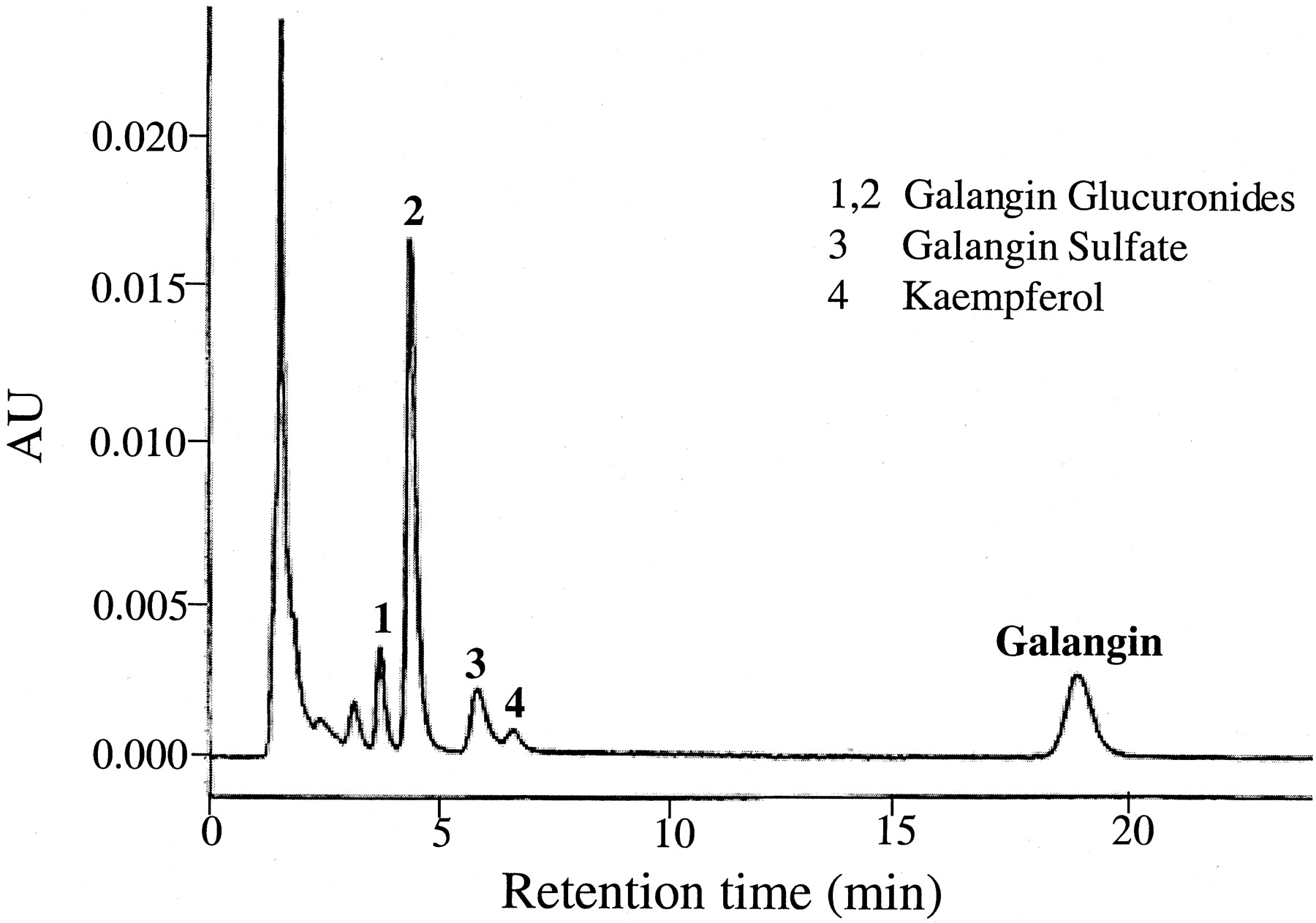
HPLC/UV analysis of a 3-h incubate of plated hepatocytes from one donor with 25 μM galangin.
The sample was analyzed directly after precipitating proteins in the medium with methanol. Peaks 1, 2, 3, and 4 are galangin metabolites. AU, absorbance units.

Metabolism of galangin by plated human hepatocytes from six donors at 1, 3, and 6 h.
△, remaining galangin; open bars, metabolite peak 1 (see Fig. 6); closed bars, metabolite peak 2; hatched bars, metabolite peak 3. Data are mean values ± S.E. of duplicate samples from six donors.
To determine the oxidative potential of the plated hepatocytes used, several experiments were conducted. CYP1A2 activity, using the ethoxyresorufin-O-deethylase assay, was 0.56 and 2.88 pmol/h/mg of cellular protein with cells from two donors. Interestingly, the hepatocytes with the higher activity were from one of the donors showing a small amount of galangin oxidation. We also homogenized hepatocytes from one of the donors. When 10 μM galangin was incubated with this homogenate, together with an NADPH-generating system, the oxidation product kaempferol could be clearly identified, based on its characteristic UV absorption and a retention time identical to reference kaempferol, as previously shown for human liver microsomes (Otake and Walle, 2002).
Discussion
In our previous study, we demonstrated that the flavonoid galangin is oxidized to kaempferol (Fig. 1) by human liver microsomes. This oxidation was catalyzed by both CYP1A2 and CYP2C9 (Otake and Walle, 2002). The apparent catalytic efficiency (Vmax/Km)) for the pooled human liver microsomes for this reaction was 13.6 μl/min/mg of protein. This is comparable to those for model substrates of CYP1A2 (e.g., phenacetin; Rodrigues et al., 1997), and CYP2C9 (e.g., tolbutamide; Miners et al., 1988), which show a reasonably high efficiency.
However, as determined in the present study, the apparentVmax/Kmvalues for the two major human liver microsomal glucuronidation reactions of galangin (i.e., peak 1 and 2 formation) were 11 to 31 times higher than for the oxidation, Table 1, using exactly the same human liver microsomes. These values are high compared with any other UGT substrates. In a recent comparison of human liver microsomal glucuronidation of 17 substrates (Soars et al., 2001), the glucuronidation of the most efficient substrate, bilirubin, is approximately 5 times lower than for galangin in our study, whereas most drugs are 2 to 3 orders of magnitude lower. This is also true for the two typical glucuronidation substrates, acetaminophen (Fisher et al., 2000) and morphine (King et al., 2000), in previous studies. Glucuronidation of steroids, such as androstanediol (Soars et al., 2001), estradiol (Fisher et al., 2000), and the bile acid, hyodeoxycholic acid (Soars et al., 2001), are glucuronidated at intermediate rates. It would be of great importance to determine what makes galangin and maybe other flavonoids such good substrates for glucuronidation.
Although the conditions for oxidation and glucuronidation of galangin were not identical, they did not markedly influence the rates. Thus, the amount of cofactors used were saturating, the linearity of the reactions were similar, and the recovery of the metabolites measured was high. Also, the different buffers used in the two reactions had no clear effect on reaction rates. The high rate of glucuronidation compared with oxidation of galangin was reflected in the small amount of microsomes needed for glucuronidation (3 μg) compared with oxidation (50 μg) to achieve initial velocity conditions.
In addition to the efficient glucuronidation of galangin by the human liver microsomes, we also showed efficient sulfate conjugation, mediated mainly by SULT1A1, although the rates could not be directly compared with oxidation and glucuronidation for this cytosolic enzyme.
To be able to get a direct comparison between the three metabolic pathways, we repeated these experiments with the S9 fraction of liver pooled from 10 donors. The conclusion was similar to that from the microsomal and cytosolic fractions (i.e., glucuronidation was 16 to 29 times more efficient than oxidation). We could now conclude that sulfation was about 7 times more efficient than the oxidation (Table3).
Consistent with the subcellular metabolism studies, we demonstrated that galangin mainly undergoes glucuronidation and sulfation in freshly plated human hepatocytes, with only trace amounts of oxidation. Fresh hepatocytes should provide a physiologically more relevant picture of the situation in vivo. Although galangin metabolism has not yet been studied in vivo, after reviewing the literature, it appears that flavonoids in general may preferentially undergo glucuronidation and sulfation [as shown for e.g., baicalin (Abe et al., 1990), liquiritigenin (Shimamura et al., 1993), diosmetin (Boutin et al., 1993), flavopiridol (Jäger et al., 1998), and quercetin (Manach et al., 1998)]. However, in neither case were intact glucuronic acid or sulfate conjugates identified. The main point is that there is little evidence that oxidation of most flavonoids may be important in vivo. However, more detailed studies in vivo in humans, preferentially using radiolabeled flavonoids, should be done.
Although the hepatocytes used in this study clearly had P450-mediated oxidative capacity, it could be argued that they had lost some of this activity after the initial plating. As galangin is metabolized by CYP1A2 and CYP2C9 (Otake and Walle, 2002), these would be the isoforms of concern. As reported previously, after 36 to 48 h in culture, as in this study, CYP1A2 activity can be expected to have declined by about 50% (Grant et al., 1987; Maurel, 1996). CYP2C9 may not have been affected this way during this rather short preincubation period (Maurel, 1996). The reason for this differential behavior appears to be that CYP1A2 is a highly inducible isoform, due to normal exposure to a variety of environmental chemicals, whereas CYP2C9 is not. The decline of CYP1A2 in culture is thus due to its slow return to noninduced conditions (Maurel, 1996). The best indicator that our cells had high activity of these isoforms was the finding that a homogenate of the cells, in the absence of cofactors for conjugation, effectively oxidized galangin to kaempferol, just as in our previous observations in human liver microsomes (Otake and Walle, 2002).
The main UGT isoform responsible for the efficient glucuronidation of galangin to form peak 1, or the 3-O-glucuronide, appears to be UGT1A9, an isoform well expressed in the human liver (Tukey and Strassburg, 2001). UGT1A9 is, however, also expressed at extrahepatic sites such as in the colon and kidney (Tukey and Strassburg, 2001). UGT1A9 was also shown to be important for the glucuronidation of the flavonoid chrysin, although UGT1A1 seemed to predominate (Walle et al., 2000a). For chrysin, which does not have a 3-hydroxyl group, the major site of glucuronidation was the 7-position (Galijatovic et al., 1999). The glucuronidation of the 7-position of galangin (i.e., peak 2), interestingly, was carried out by UGT1A9 as well as by UGT1A1 and UGT2B15. The latter finding emphasizes that we should not exclude members of the UGT2B subfamily in the glucuronidation of nonsteroid substrates. The high efficiency of glucuronidation of galangin may suggest interactions by this and other flavonoids with glucuronidation of drugs (Lautala et al., 2000), carcinogens (Ren et al., 2000), and other chemicals (Ebner et al., 1993).
In conclusion, this study provides direct evidence that galangin and maybe other flavonoids are preferentially glucuronidated and sulfated versus oxidized in hepatocytes and therefore likely also in vivo in humans. Although oxidation does occur, both glucuronidation and sulfation are more efficient. The very high rate of glucuronidation of galangin points toward potential interactions with other molecules being substrates for, in particular, UGT1A9.
Acknowledgments
We thank Kristina Walle for help with the hepatocyte experiments and in the preparation of the manuscript.
Footnotes
-
This study was supported by the National Institutes of Health Grant GM55561 and was presented in part at the Experimental Biology 2001 Meeting in Orlando, FL (March 31–April 4, 2001).
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- SULT
- sulfotransferase
- P450
- cytochrome P450
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- PAPS
- 3′-phosphoadenosine-5′-phosphosulfate
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- MS
- mass spectrometry
- Received November 6, 2001.
- Accepted February 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}