


Abstract
The objectives of this study were to evaluate the ability of 14 compounds, which differentially activate human pregnane X receptor (hPXR), to induce CYP2B6 expression and to compare CYP2B6 and CYP3A4 concentration- and time-dependent induction by select inducers. Three primary human hepatocyte preparations were treated daily for 3 days with three concentrations of all compounds. Additional concentration- and/or time-response studies were conducted with clotrimazole, phenytoin, phenobarbital, and rifampin in six preparations. CYP2B6 and CYP3A4 protein and activities were assessed by Western blotting, bupropion hydroxylation, and testosterone 6β-hydroxylation, respectively. To evaluate hPXR activation by the 14 compounds, reporter gene assays were conducted using Huh7 cells cotransfected with hPXR and a CYP2B6 (NR1)5-LUC reporter plasmid. Clotrimazole, phenobarbital, rifampin, and ritonavir strongly induced CYP2B6 and activated hPXR; dexamethasone t-butylacetate and sulfinpyrazone induced CYP2B6 weakly and activated hPXR moderately; paclitaxel strongly activated hPXR but did not increase CYP2B6 expression; carbamazepine and phenytoin moderately or strongly increased CYP2B6 expression but weakly activated hPXR; and dexamethasone, methotrexate, probenecid, sulfadimidine, and troleandomycin demonstrated weak or negligible effects on CYP2B6 and hPXR. EC50 values for CYP2B6 and CYP3A4 induction by clotrimazole, phenobarbital, phenytoin, and rifampin were strongly correlated (r2 = 0.99) and were statistically indistinguishable for clotrimazole, phenytoin, and rifampin. Kinetic constants governing time-dependent induction by phenobarbital and rifampin were also similar between CYP2B6 and CYP3A4. These results indicate that CYP2B6 is highly inducible by known CYP3A4 inducers and suggest that hPXR is a major determinant of CYP2B6-inducible expression for many, but not all, compounds evaluated in this study.
CYP2B6 has been thought historically to account for only a minor portion (<1%) of total hepatic cytochrome P450 content and to play an insignificant role in human drug metabolism (Shimada et al., 1994). However, increased interest in this enzyme has been stimulated by the discovery of polymorphic and ethnic differences in CYP2B6 expression (Ariyoshi et al., 2001; Lang et al., 2001), identification of additional substrates for CYP2B6, (http://www.gentest.com/human_p450_database/index.html), and evidence for cross-regulation with CYP3A4 expression (Sueyoshi et al., 1999; Moore et al., 2000; Goodwin et al., 2001; Mäkinen et al., 2002). In contrast to earlier investigations, recent studies report higher expression levels of CYP2B6 in human liver microsomes using more sensitive and specific immunochemical and biochemical detection methods. These studies also report large interindividual differences in hepatic CYP2B6 protein levels ranging from 20- to 250-fold (Code et al., 1997; Ekins et al., 1998; Stresser and Kupfer, 1999; Hanna et al., 2000; Hesse et al., 2000). In addition, CYP2B6 activities have been reported to vary from 25-fold using S-mephenytoin as substrate (Ekins et al., 1998) to 80-fold using bupropion as substrate (Faucette et al., 2000). These interindividual differences in CYP2B6 catalytic capacity would be expected to result in variable systemic exposure, and perhaps response, to drugs metabolized by CYP2B6, including the antineoplastics cyclophosphamide and ifosfamide (Roy et al., 1999); the antiretrovirals nevirapine (Erickson et al., 1999) and efavirenz (Du et al., 2000); the anesthetics propofol (Court et al., 2001) and ketamine (Yanagihara et al., 2001); and the anti-Parkinsonian selegiline (Hidestrand et al., 2001).
Despite the potential clinical impact of CYP2B6 induction, there is relatively little information available regarding the chemical diversity, efficacy, and potency of human CYP2B6 inducers. Several studies have demonstrated increased CYP2B6 expression by CYP3A4 inducers, such as dexamethasone, cyclophosphamide, phenobarbital, rifampin, troglitazone, and calcium channel antagonists (Chang et al., 1997; Gervot et al., 1999; LeCluyse et al., 2000; Sahi et al., 2000; Goodwin et al., 2001; Drocourt et al., 2001). However, these studies examined a limited number of inducers and inducer concentrations, evaluated CYP2B6 expression at the level of mRNA only, and/or used nonselective probes of CYP2B6 protein and catalytic activity. Currently, a more thorough characterization of CYP2B6 induction potential at the level of activity is feasible because of the recent validation of bupropion as a selective substrate probe for this enzyme (Faucette et al., 2000; Hesse et al., 2000).
In contrast to human CYP2B6, induction of rodent CYP2B enzymes has been well characterized (Waxman and Azaroff, 1992; Honkakoski et al., 1998a). Studies by Negishi and colleagues have established that activation and/or translocation of the constitutive androstane receptor (CAR)1 mediates induction of rodent CYP2B genes by phenobarbital (PB)-type inducers (Honkakoski et al., 1998b; Kawamoto et al., 1999). However, notable species differences in CAR activation have limited the extrapolation of these findings to humans (Moore et al., 2000). To date, no compound has been identified that significantly activates human CAR to a degree consistent with levels of CYP2B6 induction observed in human hepatocytes. In contrast, hPXR, recognized as the primary regulator of CYP3A4 expression (Bertilsson et al., 1998; Lehmann et al., 1998), has been shown to mediate transactivation of the CYP2B6 phenobarbital-responsive enhancer module (PBREM) in response to rifampin (Goodwin et al., 2001). Several laboratories have demonstrated that both human CAR and PXR can bind and activate response elements in the promoter regions of CYP2B6 and CYP3A4 genes (Goodwin et al., 2001; Mäkinen et al., 2002; Wang et al., 2003b). Consequently, prototypical inducers may differentially activate single or multiple nuclear receptors to induce different P450 genes via common or distinct response elements. In contrast to rodent systems, it is conceivable that known PXR activators and CYP3A inducers may modulate CYP2B6 expression in human liver.
The primary objectives of this study were to characterize the ability of 14 compounds to induce CYP2B6 protein and catalytic activity in primary cultures of human hepatocytes and activate hPXR in CYP2B6 reporter gene assays. Most of these compounds have been demonstrated previously to induce hepatic CYP3A4 differentially in vitro or in vivo. An additional objective was to compare the concentration- and time-dependent profiles of CYP2B6 and CYP3A4 induction by a subset of these compounds.
Materials and Methods
Chemicals. Bupropion hydrochloride, dimethyl sulfoxide (DMSO), and triethylamine were purchased from Sigma-Aldrich (St. Louis, MO). Analytical grade acetonitrile and methanol were purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). Testosterone and 6β-hydroxytestosterone were purchased from Steraloids (Newport, RI). Hydroxybupropion was obtained from Glaxo-SmithKline, Inc. (Research Triangle Park, NC). DuPont Pharmaceuticals, Inc. (Wilmington, DE) kindly supplied the test compounds for induction studies [carbamazepine (CMZ), clotrimazole (CLZ), dexamethasone (DEX), dexamethasone t-butylacetate (DTBA), methotrexate (MTX), paclitaxel (PAX), PB, phenytoin (PHN), probenecid (PROB), rifampin (RIF), ritonavir (RIT), sulfadimidine (SDM), sulfinpyrazone (SPZ), and troleandomycin (TAO)]. All other chemicals were of the highest grade commercially available.
Biological Reagents. Dulbecco's modified Eagle's medium (DMEM) and DMEM/F12 medium were purchased from Invitrogen (Carlsbad, CA). Polyclonal rabbit anti-human CYP2B6 and CYP3A4 antibodies were purchased from Chemicon International, Inc. (Temecula, CA), while alkaline phosphatase- or horseradish peroxidase-conjugated goat anti-rabbit IgG was purchased from Valeant Pharmaceuticals International (Costa Mesa, CA). Charcoal-stripped, dextran-treated fetal bovine serum was acquired through Hyclone Laboratories (Logan, UT). Effectene transfection reagent was obtained from QIAGEN (Valencia, CA). Dual-Luciferase Reporter Assay System was purchased from Promega (Madison, WI). Other biological reagents were purchased from commercial suppliers and were American Chemical Society or molecular biology grade.
Plasmids. The full-length cDNAs for hPXR and hCAR were subcloned into pCMV · SPORT and pCR3 vectors (Invitrogen), respectively. The human glucocorticoid receptor α (pCMVhGRα) expression plasmid, a kind gift from Dr. John Cidlowski (National Institute of Environmental Health Sciences, Research Triangle Park, NC), was constructed as previously described (Oakley et al., 1996). A CYP2B6 (NR1)5-LUC plasmid was generated by cloning a 5-repeat human NR1 sequence (5′-gatcACTGTACTTTCCTGACCTTgatc-3′) into the KpnI/XhoI site of pGL3-promoter vector (Promega).
Isolation and Culture of Human Hepatocytes. Human liver tissues were obtained as surgical waste or rejected donor livers from the University of North Carolina at Chapel Hill or Incara Cell Technologies (Research Triangle Park, NC). All hepatic tissues were procured by qualified medical staff following donor consent and with approval from the appropriate hospital ethics committees. Hepatocytes were isolated by a modification of the two-step collagenase digestion method as previously described (Hamilton et al., 2001). Table 1 provides the age, gender, ethnicity, and medical information for the nine donors included in this study. Isolated hepatocytes were cultured according to methods described by LeCluyse et al. (2000). Cultures were maintained for 36 to 48 h before initiating treatment with test compounds.
Characteristics of human liver donors
Treatment of Human Hepatocyte Cultures. Initially, hepatocyte cultures from donors HL-N95, HL-N98, and HL-N100 were treated daily for 3 days with solvent (0.1% DMSO) or 14 test compounds at final concentrations of 2, 10, and 20 μM (except for PB, which was tested at 50, 150, and 250 μM). For detailed concentration-response studies, hepatocytes from five donors were treated daily for 3 days with solvent (0.1% or 0.25% DMSO), CLZ (0.4–100 μM), PB (10 μM to 2 mM), PHN (0.4–500 μM), or RIF (0.1–250 μM). Three preparations (HL-N131, HL-N140, and HL-N145) were exposed to all four inducers, whereas one preparation each was exposed to RIF and PB only (HL-N147) or CLZ and PHN only (HL-N148). In time course experiments, hepatocytes from donor HL-N154 were treated for 0, 24, 48, 72, 96, or 120 h with 0.1% DMSO, 10 μM RIF, or 1 mM PB. For cultures with cumulative treatment times longer than 24 h, fresh medium containing solvent or drug was prepared daily and replaced every 24 h. At the end of each treatment period, cells were harvested and microsomes were prepared as previously described (Hamilton et al., 2001). Homogenate and microsomal protein concentrations were determined with the BCA Protein Assay kit using bovine serum albumin as standard (Pierce Chemical, Rockford, IL).
Determination of CYP2B6 and CYP3A4 Microsomal Activities. CYP2B6 and CYP3A4 activities were measured in duplicate using microsomes isolated from cultured human hepatocytes using bupropion and testosterone as respective catalytic probes. Rates of formation of hydroxybupropion from bupropion (500 μM) were determined using methods previously described by Faucette et al. (2000), while rates of formation of 6β-hydroxytestosterone from testosterone (250 μM) were determined with an assay previously described by Pearce et al. (1996). The results of CYP3A4 determinations for donors HL-N95, HL-N98, and HL-N100 have been reported in a prior publication (Luo et al., 2002).
Concentrations of hydroxybupropion were determined by reverse-phase high performance liquid chromatography with UV detection at a wavelength of 210 nm. Separation of peaks was achieved at ambient temperature using a 5-μm 15 × 0.39 cm Hypersil BDS C18 column preceded by a C18 guard cartridge. Elution was achieved at 1 ml/min using a binary gradient of solvent A (0.025 M potassium phosphate, 1 ml/l triethylamine, pH 7.0, in 95:5 v/v water/acetonitrile) and solvent B (acetonitrile). Concentrations of 6β-hydroxytestosterone were quantified using a previously described reversephase high performance liquid chromatography assay (Pearce et al., 1996).
Western Immunoblot Analysis. Homogenate or microsomal protein (2.5–5 μg for CYP3A4 and 15–35 μg for CYP2B6) was subjected to SDS-polyacrylamide gel electrophoresis (9%) and transferred onto an Immobilon-P polyvinylidene difluoride membrane (Millipore Corporation, Bedford, MA). Membranes were probed with polyclonal rabbit CYP2B6- or CYP3A4-specific antibodies at dilutions of 1:5000 or 1:8000, respectively, followed by goat anti-rabbit alkaline phosphatase- or horseradish peroxidase-conjugated anti-bodies at dilutions of 1:800. Blots were developed by colorimetric detection using 5-bromo-4-chloroindolyl-phosphatase/nitrobenzotetrazolium (BCIP/NBT) (Kirkegaard and Perry Laboratories, Gaithersburg, MD) or by using enhanced chemiluminescence detection reagent (Amersham Biosciences Inc., Piscataway, NJ). The relative amounts of CYP2B6 or CYP3A4 protein in select blots were estimated by densitometric analysis using Quantity One software (Bio-Rad, Hercules, CA).
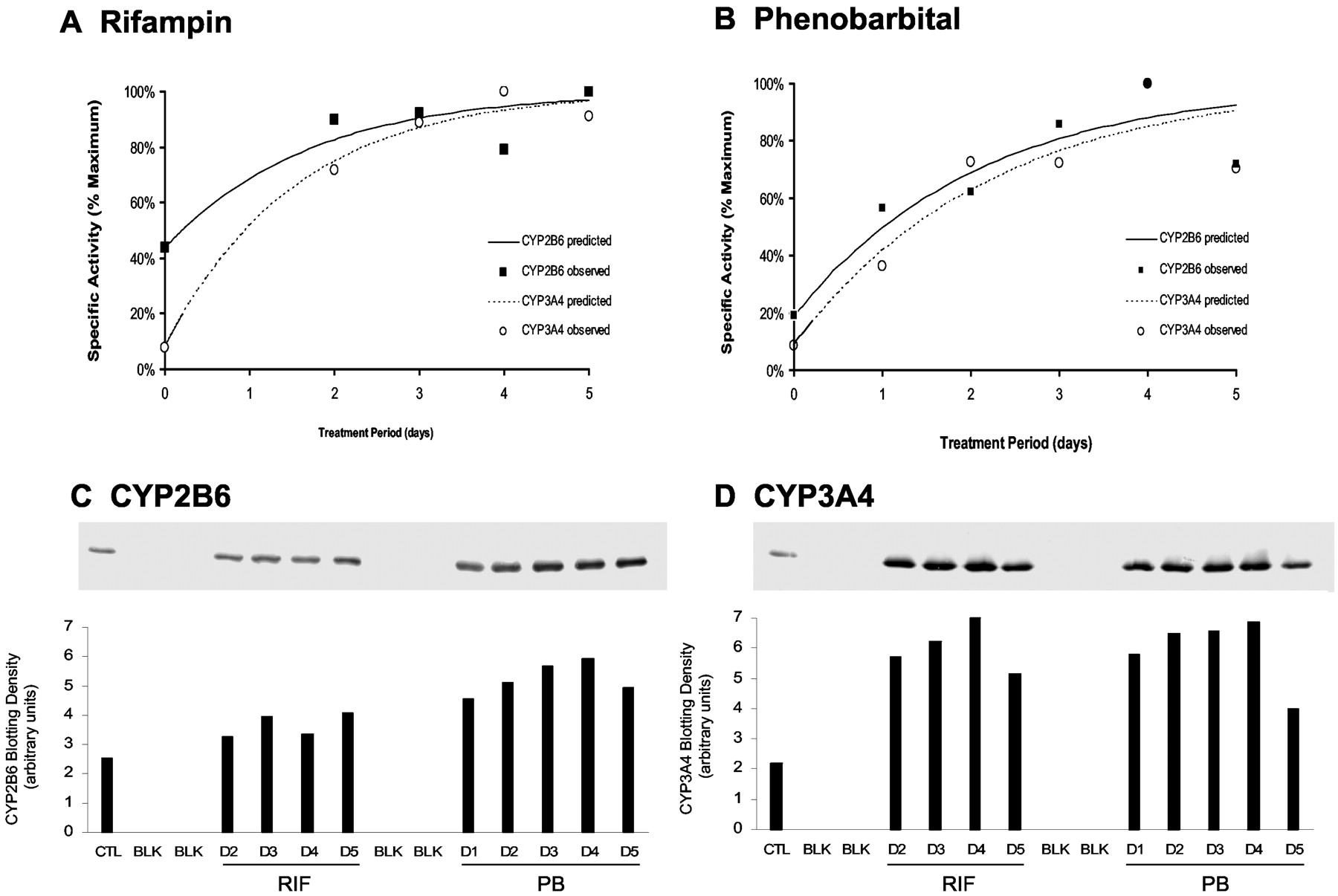
Modeling of Potency and Time Course of Enzyme Induction. Concentration-effect profiles were generated for induction of CYP2B6 and CYP3A4 expression by CLZ, PHN, PB, and RIF. CYP2B6 and CYP3A4 catalytic activities were converted to percentage of maximum [E (%)] according to the equation  where Eobs is the observed activity, E0 is the baseline activity, and Emax is the maximum activity achieved. The relationship between E (%) for enzyme activity and inducer concentration was fit with a standard Hill equation
where Eobs is the observed activity, E0 is the baseline activity, and Emax is the maximum activity achieved. The relationship between E (%) for enzyme activity and inducer concentration was fit with a standard Hill equation 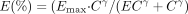 by nonlinear least-squares regression to recover estimates of the EC50 and sigmoidicity factor (γ) for each concentration-effect profile. Because the model equation was fit only to concentration-effect data consistent with induction and a sigmoidal pattern of response, enzyme activities at the highest evaluated concentrations that were less than 80% of Emax were excluded from analysis. Final parameter estimates obtained by regression analyses were substituted into the Hill equation to simulate percentage of maximum enzyme activity over the range of inducer concentrations tested in human hepatocyte cultures.
by nonlinear least-squares regression to recover estimates of the EC50 and sigmoidicity factor (γ) for each concentration-effect profile. Because the model equation was fit only to concentration-effect data consistent with induction and a sigmoidal pattern of response, enzyme activities at the highest evaluated concentrations that were less than 80% of Emax were excluded from analysis. Final parameter estimates obtained by regression analyses were substituted into the Hill equation to simulate percentage of maximum enzyme activity over the range of inducer concentrations tested in human hepatocyte cultures.
CYP2B6 or CYP3A4 activity versus treatment time data from RIF- and PB-treated hepatocytes (HL-N154) were fit with a first-order exponential approach to plateau by nonlinear least-squares regression using the equation  where K is the first-order rate constant. Final estimates of K recovered by regression analyses were substituted into the above equation to simulate percentage of maximum enzyme activity over the entire time course of RIF or PB treatment.
where K is the first-order rate constant. Final estimates of K recovered by regression analyses were substituted into the above equation to simulate percentage of maximum enzyme activity over the entire time course of RIF or PB treatment.
Transfection of Hepatoblastoma Huh7 Cells. Human liver-derived Huh7 cells were cultured in DMEM supplemented with 10% charcoal-stripped fetal bovine serum and antibiotics. Cells were seeded into 24-well plates at 5 × 104 cells/well and transfected 24 h later with Effectene transfection reagent (QIAGEN). Transfection mixes were prepared in DMEM/F12 containing 0.1 μM dexamethasone and consisted of 100 ng of CYP2B6 (NR1)5 firefly luciferase reporter plasmid, 25 ng of hPXR or hCAR expression plasmid, 25 ng of hGRα expression plasmid, and 10 ng of Renilla luciferase reporter vector (pRL-TK) as internal control (Wang et al., 2003a). Twenty-four hours after transfection, cells were treated for 24 h in the presence of 0.1 μM dexamethasone and solvent (0.1% DMSO) or test compounds (20 μM, except 250 μM and 500 μM for PB and 100 μM additionally for PHN). Luciferase activities were measured with the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega). Reporter activities were determined from three independent transfections and calculated as the ratio of firefly luciferase to Renilla luciferase activities and expressed relative to control.
Statistical Analyses. EC50 values for induction of CYP2B6 and CYP3A4 by select inducers were compared using a two-tailed paired t test. Orthogonal linear regression was used to evaluate the relationship between EC50 values for CYP2B6 induction and EC50 values for CYP3A4 induction. Significance of the correlations was determined by an F test. A probability of less than 0.05 was used as the criterion for statistical significance.
Results
Induction of CYP2B6 Protein and Activity in Primary Human Hepatocyte Cultures. Human hepatocytes were treated daily for 3 days with 14 clinically used drugs that have been reported to cause weak, moderate, or strong induction of CYP3A4 (Luo et al., 2002 and references therein). CYP2B6 catalytic activities were determined by measuring bupropion hydroxylation in microsomal samples from hepatocyte cultures. Figure 1A demonstrates the mean concentration-dependent effects of these 14 compounds on CYP2B6 catalytic activities obtained from three preparations of primary human hepatocytes (HL-N95, HL-N98, and HL-N100). Table 2 provides fold induction values for individual livers and demonstrates interdonor variability in the extent of CYP2B6 induction. Based on mean fold induction values, CLZ, PHN, PB, and RIF were classified as strong inducers (≥6-fold), CMZ and RIT as moderate inducers (4- to 6-fold), and DTBA as a weak inducer (2-fold) of CYP2B6 activity. Using the same criteria, ritonavir alternatively can be classified as a strong inducer (7.1-fold) and sulfinpyrazone (2.2-fold) as a weak inducer, based on data obtained in a single hepatocyte preparation at the highest concentration tested. For strong inducers, the maximum levels of induction observed in individual human livers ranged from 7- to 20-fold for CLZ, 6- to 10-fold for PHN, 6- to 7-fold for PB, 7- to 10-fold for RIF, and 4- to 7-fold for RIT. No significant induction (<2-fold) of CYP2B6 activity was observed at any concentration of DEX, MTX, PAX, PROB, SDM, and TAO.
Concentration-dependent induction of CYP2B6 (A) and CYP3A4 (B) activities by 14 compounds in human hepatocytes.
A, rates of bupropion hydroxylation were determined as described under Materials and Methods using microsomes isolated from primary cultures of human hepatocytes treated daily for 3 days with solvent (0.1% DMSO) or 2, 10, and 20 μM concentrations of test compounds (except 50, 150, and 250 μM for PB). Rates of formation represent the mean and S.D. from three human hepatocyte preparations (HL-N95, HL-N98, and HL-N100). B, rates of testosterone 6β-hydroxylation for HL-N95, HL-N98, and HL-N100 have been reported in terms of fold induction in a previous publication by Luo et al. (2002), but are reported here as specific activities (pmol/min/mg protein, mean ± S.D.) for comparison with CYP2B6 activities. CTL, control.
Interdonor variability in induction of CYP2B6 activity in primary human hepatocyte cultures Rates of bupropion hydroxylation were determined in duplicate in microsomes isolated from three human hepatocyte preparations (HL-N95, HL-N98, HL-N100) treated daily for 3 days with the indicated concentrations of tests compounds. Bupropion hydroxylase activities in control samples were 23, 16, and 45 pmol/min/mg protein for HL-N95, HL-N98, and HL-N100, respectively. Values in the table represent fold induction over control activities.
Microsomes prepared from a representative hepatocyte preparation (HL-N95) were subjected to Western blot analysis using anti-CYP2B6 antibodies (Fig. 2). CYP2B6 protein was only weakly detectable in control cultures, consistent with the low constitutive expression of this enzyme in human liver. However, CYP2B6 immunoreactive protein increased in a concentration-dependent manner for CMZ, CLZ, DTBA, PB, PHN, RIF, and RIT. No discernible changes in CYP2B6 protein expression were observed in microsomal samples from hepatocytes treated with DEX, MTX, PAX, PROB, SDM, SPZ, and TAO. In general, the patterns of increase in CYP2B6 protein levels were consistent with those observed for activities for HL-N95 and the other hepatocyte preparations (data not shown).

Western blot of concentration-dependent induction of CYP2B6 protein by 14 compounds in human hepatocytes.
Equal amounts of microsomal protein (20 μg) from HL-N95 were subjected to SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and probed with polyclonal rabbit anti-human CYP2B6 antibodies followed by alkaline phosphatase-conjugated goat anti-rabbit IgG. Blots were developed using BCIP/NBT as substrate solution. CTL, control; BLK, blank.
Comparison between CYP2B6 and CYP3A4 Induction. The concentration-dependent effects of the 14 compounds on CYP3A4 activities were determined by measuring testosterone 6β-hydroxylation with microsomes isolated from hepatocyte cultures HL-N95, HL-N98, and HL-N100 (Fig. 1B). Although fold induction of CYP3A4 activities by the 14 compounds was reported for these hepatocyte preparations in a previous publication (Luo et al., 2002), CYP3A4 specific activities (pmol/min/mg protein) are shown in Fig. 1B for comparison with CYP2B6 activities. Mean fold induction values for CYP3A4 were lower than for CYP2B6 due to higher basal CYP3A4 activities in control cultures. With the exception of DEX, all compounds identified as strong, moderate, or weak inducers of CYP3A4 activity (≥1.5 fold) in the previous study (Luo et al., 2002) were shown to induce CYP2B6 activity by at least 2-fold in the present study. MTX, PAX, PROB, SDM, and TAO induced neither CYP3A4 nor CYP2B6 activities. In addition, compounds shown to activate hPXR to varying degrees in previous studies (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998; Jones et al., 2000; Moore et al., 2000; Luo et al., 2002) were found to induce CYP2B6 expression in this study, with the exception of DEX, PAX, SDM, and TAO. These compounds activated PXR moderately or strongly (>5-fold) in CYP3A4 reporter gene assays (Lehmann et al., 1998; Luo et al., 2002) but failed to induce CYP2B6 in human hepatocyte cultures. However, PAX, SDM, and TAO also failed to significantly increase CYP3A4 activity (Fig. 1B; Luo et al., 2002).
Concentration-Effect Profiles for CYP2B6 and CYP3A4 Induction by CLZ, PHN, PB, and RIF. Because initial studies demonstrated a qualitative correlation between CYP2B6 and CYP3A4 induction by most test compounds, additional studies were performed with strong CYP2B6 inducers over a broader range of concentrations to compare the concentration-effect relationships for CYP2B6 and CYP3A4 induction. Measures of CYP2B6 and CYP3A4 expression were determined with microsomes isolated from five different human hepatocyte preparations (HL-N131, HL-N140, HL-N145, HL-N147, and HL-N148) treated daily for 3 days with CLZ (0.4–100 μM), PB (10 μM to 2 mM), PHN (0.4–500 μM), and RIF (0.1–250 μM). Preliminary experiments revealed that concentrations greater than the maximum concentrations in the ranges above were associated with compound insolubility in culture media or overt cytotoxicity in most hepatocyte preparations.
Representative concentration-response curves for RIF, PB, PHN, and CLZ induction of CYP2B6 and CYP3A4 activities are shown in Fig. 3, A-D, respectively. The concentration-dependent effects of these inducers on CYP2B6 and CYP3A4 immunoreactive proteins are shown in Fig. 4, A-D, for HL-N131. In general, the patterns of concentration-dependent increases in CYP2B6 and CYP3A4 protein levels correspond with the increases in bupropion hydroxylation and testosterone 6β-hydroxylation. For most hepatocyte preparations, maximum induction (Emax) of CYP2B6 activities was observed at 250 μM or 400 μM for PHN, 20 μM or 40 μM for CLZ, and 2 mM for PB. CYP3A4 activities were induced maximally at 250 μM or 400 μM for PHN, 20 μM for CLZ, and 2 mM for PB. Although maximum increases in both CYP2B6 and CYP3A4 activities were observed at 100 μM for RIF, relatively small increases in activities occurred between 10 μM and 100 μM for CYP2B6 and 2 μM to 100 μM for CYP3A4.

Representative concentration-effect profiles for induction of CYP2B6 and CYP3A4 activities by prototypical inducers.
Rates of bupropion hydroxylation and testosterone 6β-hydroxylation were determined with microsomes isolated from human hepatocytes (HL-N140, HL-N145, HL-N147, and HL-N148) exposed daily for 3 days to solvent (0.1% DMSO), CLZ (0.4–40 μM), PB (10 μM to 2 mM), PHN (0.4–400 μM), and RIF (0.1–150 μM). The Hill equation was fit to CYP2B6 and CYP3A4 concentration-rate data by nonlinear regression to obtain estimates of the EC50 values for each concentration-effect profile. Final parameter estimates were substituted into the Hill equation to simulate percentage of maximum enzyme activity over the range of tested inducer concentrations. The concentration-effect profiles shown for RIF (A) and PB (B) represent data from HL-N140, and those for PHN (C) and CLZ (D) represent data from HL-N148. Symbols in the graphs represent observed data, whereas dashed or solid lines indicate predicted data obtained from the model fit.

Representative Western blots for concentration-dependent induction of CYP2B6 and CYP3A4 proteins by prototypical inducers.
Cultured hepatocytes from donor HL-N131 were exposed daily to CLZ (0.4–100 μM), PB (10 μM to 2 mM), PHN (0.4–500 μM), and RIF (0.1–250 μM) for 3 days. Homogenate protein isolated from these hepatocytes were analyzed by Western immunoblotting with CYP2B6 (A and C) and CYP3A4 (B and D) polyclonal antibodies as described under Materials and Methods. The CYP2B6 band is indicated by an arrow for those blots with more than one visible band per lane. CTL, control.
Table 3 lists the mean ± S.D. values of EC50 estimates for CYP2B6 and CYP3A4 induction by these compounds obtained in three human livers (HL-N140, HL-N145, and HL-N147 for RIF and PB; and HL-N140, HL-N145, and HL-N148 for PHN and CLZ) with sufficient data points to ensure unbiased pharmacodynamic modeling. The EC50 values for CYP2B6 induction by RIF, PHN, CLZ, and PB ranged from 0.68 to 1.7 μM, 23 to 47 μM, 2.4 to 14 μM, and 188 to 293 μM, respectively. The ranges of the corresponding values for CYP3A4 induction were 0.27 to 0.47 μM for RIF, 9.9 to 23 μM for PHN, 5.0 to 5.8 μM for CLZ, and 106 to 178 μM for PB. When results from these human hepatocyte cultures were pooled, there were no statistically significant differences between the EC50 values for induction of CYP2B6 and CYP3A4 expression by CLZ, PHN, or RIF (Table 3). As expected, a strong correlation was observed for the relationship between the potency of CYP2B6 and CYP3A4 induction by CLZ, PHN, PB, and RIF (r2 = 0.99, p < 0.005). However, the slope of the line describing the relationship between CYP2B6 and CYP3A4 induction potencies was not unity, suggesting that these enzymes are regulated differentially to some degree in a compound-specific fashion. This premise is supported also by the statistically different EC50 values for CYP2B6 and CYP3A4 induction by PB (Table 3).
Comparison of EC50 values for concentration-dependent induction of CYP2B6 and CYP3A4 activities by prototypical inducers Rates of bupropion hydroxylation and testosterone 6β-hydroxylation were determined with microsomes isolated from human hepatocyte preparations treated daily for 3 days with 0.4 to 40 μM CLZ, 10 μM to 2 mM PB, 0.4 to 400 μM PHN, and 0.1 to 150 μM RIF. The relationship between enzyme activity and inducer concentration was fit with the Hill equation by nonlinear regression to recover estimates for the EC50 and sigmoidicity factor (γ). The EC50 values represent the mean ± S.D. from three individual livers (HL-N140, HL-N145, and HL-N147 for RIF and PB, and HL-N140, HL-N145, and HL-N148 for PHN and CLZ). Statistical significance of the differences in the EC50 values for CYP2B6 and CYP3A4 induction was determined using a two-tailed paired t test.
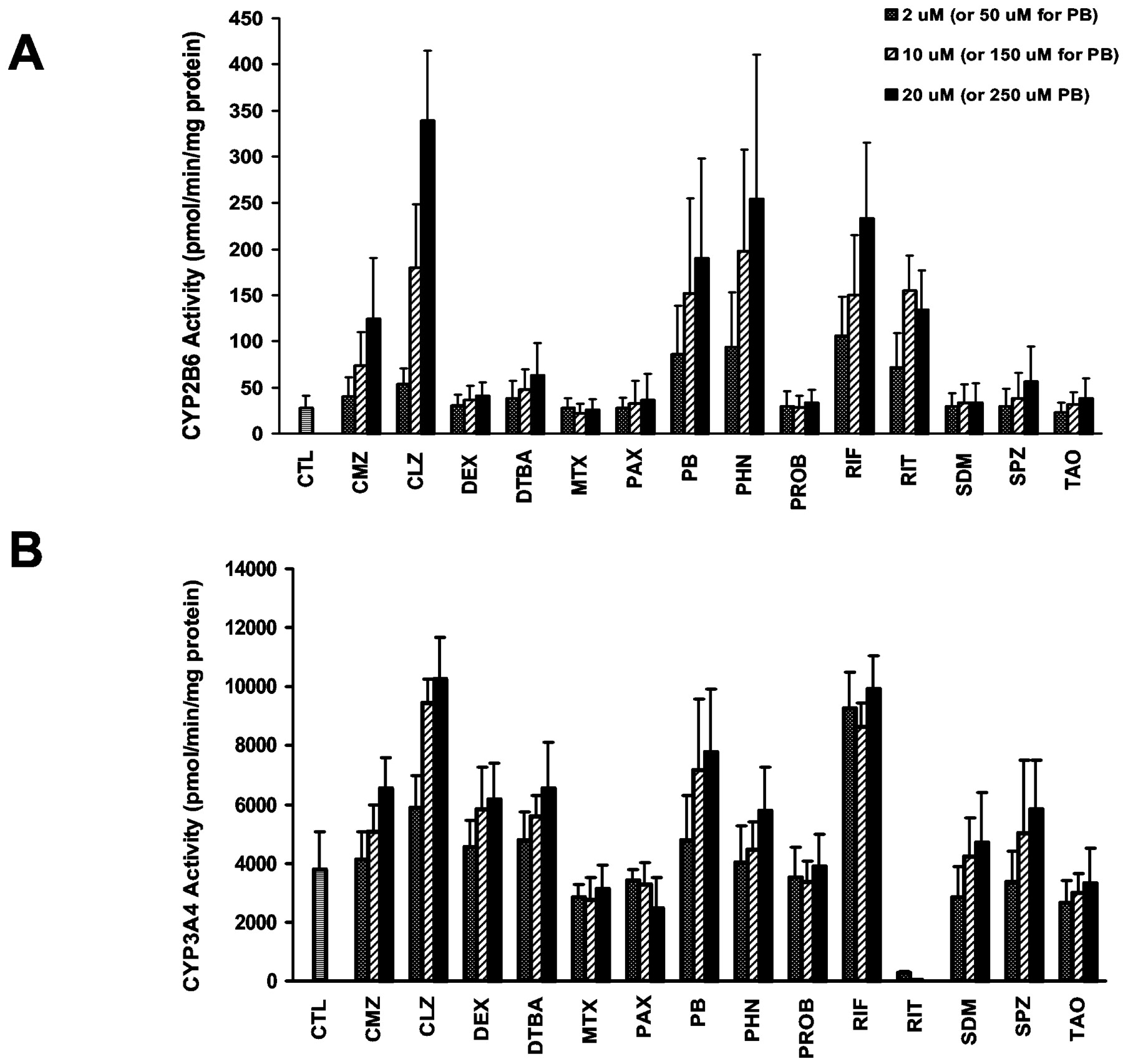
Temporal Profiles of CYP2B6 and CYP3A4 Induction. Hepatocytes from donor HL-N154 were exposed to 10 μM RIF and 1 mM PB for various times to compare the temporal kinetics of CYP2B6 and CYP3A4 induction. At all treatment periods, CYP2B6 activities were higher from PB-treated hepatocytes compared with those from RIF-treated hepatocytes, whereas the opposite was observed for CYP3A4 activities (data not shown). As shown in Fig. 5, A and B, the temporal profiles of RIF and PB induction of CYP2B6 activities were similar to those of CYP3A4 when activities were expressed in terms of percentage maximum. For RIF, CYP2B6 and CYP3A4 activities reached an approximate plateau within 48 h of treatment, with only minor changes in activities observed with an additional 48 to 72 h of treatment. In contrast, CYP2B6 and CYP3A4 activities in human hepatocytes treated with PB reached a plateau with 96 h of treatment, followed by a slight decline in activities after 120 h of treatment. The time-dependent changes in CYP2B6 and CYP3A4 protein expression in the microsomes from RIF- and PB-treated hepatocytes were similar to those observed for activities (Fig. 5, C and D). Mean ± S.E. values of the first-order rate constants for PB induction of CYP2B6 and CYP3A4 activities were 0.48 ± 0.10 day-1 and 0.45 ± 0.09 day-1, respectively, whereas those for RIF induction of CYP2B6 and CYP3A4 were 0.59 ± 0.17 day-1 and 0.65 ± 0.07 day-1, respectively.
Time-dependent induction of CYP2B6 and CYP3A4 expression by PB and RIF.
Human hepatocytes from donor HL-N154 were exposed to solvent (0.1% DMSO), 1 mM PB, or 10 μM RIF for 0, 24, 48, 72, 96, and 120 h. For cultures with cumulative treatment times longer than 24 h, fresh medium containing solvent or drug was prepared daily and replaced every 24 h. CYP2B6 and CYP3A4 activities were measured with microsomal samples from hepatocytes treated with RIF (A) or PB (B). To account for differences in the magnitude of rates of bupropion hydroxylation and testosterone 6β-hydroxylation and allow relative comparison between CYP2B6 and CYP3A4, microsomal activities were expressed as a percentage of the maximum value. The equation E = E0 + (Emax - E0)(1 - e-[supi]K) was fit to time-dependent data using nonlinear regression to recover estimates of the first-order rate constant K. In A and B, symbols represent observed data, whereas dashed and solid lines indicate predicted data obtained from the model fit. The corresponding effects on CYP2B6 and CYP3A4 protein are shown in C and D, respectively. CTL, control; BLK, blank.
Correlation between CYP2B6 Induction and hPXR and hCAR Activation of CYP2B6 Reporter Gene Expression. As described above, most compounds found to activate hPXR in CYP3A4 reporter gene assays (Luo et al., 2002) were shown to induce CYP2B6 expression in primary human hepatocyte cultures. The -1.7-kilobase promoter region of the CYP2B6 gene contains a 51-base pair PBREM consisting of two imperfect DR4 motifs (NR1 and NR2). Recent studies indicate that hPXR can also bind to the NR1 and NR2 motifs within the PBREM element (Xie et al., 2000; Goodwin et al., 2001; Mäkinen et al., 2002). Although both the NR1 and NR2 are required for optimal activation of CYP2B genes, the NR1 sequence is considered the most quantitatively important motif within the PBREM (Honkakoski et al., 1998b; Sueyoshi et al., 1999; Goodwin et al., 2001).
To obtain an enhanced understanding of the relationship between CYP2B6 induction and hPXR or hCAR activation, transfection assays were conducted using reporter constructs containing five copies of the NR1 motif within the CYP2B6 PBREM. Assays were performed with Huh7 cells cotransfected with expression plasmids for hPXR or hCAR in the presence of the 14 compounds previously tested for their CYP2B6 and CYP3A4 induction potentials. An expression plasmid for hGRα was transfected also because data from this laboratory and others suggest that dexamethasone-activated GR can enhance CYP2B6 induction and reporter activity in the presence of PXR and CAR activators (Pascussi et al., 2000; Wang et al., 2003a). Notably, in the presence of the 14 compounds, CYP2B6 reporter gene expression was not increased above control by transfection of hGRα alone (data not shown).
Results from the transfection experiments utilizing the CYP2B6 (NR1)5-LUC reporter plasmid with hPXR and hGRα are shown in Fig. 6. Based on relative luciferase activities, CLZ, PB, PAX, RIF, and RIT were classified as strong activators (>10-fold); DTBA, 100 μM PHN, and SPZ as moderate activators (5- to 10-fold); and DEX and 20 μM PHN (∼1.5-fold) as weak activators of hPXR. CMZ, MTX, PROB, SDM, and TAO demonstrated negligible effects on hPXR activity. For CLZ, DEX, MTX, PB, PROB, RIF, RIT, SDM, and TAO, correspondence was observed between the degree of PXR activation (or lack thereof) in CYP2B6 reporter gene assays and the degree of CYP2B6 induction in primary human hepatocyte cultures. However, discrepancies were noted between the magnitudes of hPXR activation and CYP2B6 induction for CMZ, DTBA, PAX, PHN, and SPZ. Although PAX was the third strongest hPXR activator, CYP2B6 protein and activity were unchanged by its presence in human hepatocyte cultures. In contrast, DTBA and SPZ induced CYP2B6 in all or some hepatocyte preparations, but to a lesser degree than predicted from their hPXR activation potentials. Conversely, CMZ and PHN resulted in moderate or strong CYP2B6 induction, but negligible or relatively low hPXR activation. Even at a higher PHN concentration (100 μM), relatively modest PXR activation was observed compared with the other strong CYP2B6 inducers.

Activation of CYP2B6 (NR1)5-LUC reporter gene by human PXR and GR in Huh7 cells.
A luciferase reporter vector containing five repeats of the NR1 motif within the CYP2B6 PBREM was cotransfected with hPXR, hGRα, and pGL-TK (as internal control) in Huh7 cells. Transfected cells were treated with 20 μM concentrations of the 14 compounds (or 250 μM and 500 μM for PB and 100 μM additionally for PHN) or solvent (0.1% DMSO), in the presence of 0.1 μM DEX for 24 h before dual luciferase measurement. Data represent the mean ± S.D. of three individual transfections. CTL, control.
The discordance between the magnitudes of increase in CYP2B6 expression and CYP2B6 reporter gene activity by CMZ and PHN suggests that these inducers may regulate CYP2B6 gene expression through alternative nuclear receptor pathways, such as hCAR. Accordingly, the 14 compounds were screened for their ability to activate this nuclear receptor in CYP2B6 reporter gene assays. Because of the high constitutive activity of hCAR in cell-based transfection assays (Kawamoto et al., 1999), which precludes unequivocal identification of hCAR ligands, no increases in CYP2B6 reporter gene activities were predicted for any of the 14 compounds. However, the potential for the 14 compounds to activate hCAR was evaluated nonetheless because of the possibility that its high constitutive activation would be modulated by cotransfection of hGRα. As predicted, none of the cultures treated with the 14 compounds exhibited increased CYP2B6 reporter gene activity above that of control cultures (data not shown). By contrast, reporter gene activities were reduced by approximately 50% for CLZ, PAX, RIT, and SDM, suggesting repression of the constitutive activity of hCAR by these compounds (data not shown).
Discussion
Primary cultures of human hepatocytes were used in this study to demonstrate induction of CYP2B6 expression by a diverse array of known CYP3A4 inducers, including CMZ, PB, PHN, CLZ, RIF, RIT, DTBA, and SPZ. The highly inducible nature of CYP2B6 may contribute in part to the large interindividual variability in hepatic CYP2B6 expression and to clinically significant drug interactions involving CYP2B6 substrates. The levels of CYP2B6 induction observed in hepatocyte cultures are consistent with previous in vivo observations of altered pharmacokinetics of CYP2B6 substrates in the presence of prototypical inducers. For example, increased clearance and decreased systemic exposure of bupropion, cyclophosphamide, ifosfamide, and nevirapine have been observed upon concurrent administration of the inducers CMZ, PHN, or RIF (Ketter et al., 1995; Ducharme et al., 1997; Williams et al., 1999; Ribera et al., 2001). These examples suggest that drug interactions originally attributed to selective CYP3A4 induction can be explained alternatively by concurrent CYP2B6 induction.
Delineation of the molecular mechanism(s) underlying CYP2B6 induction would facilitate high throughput screening of new chemical entities for CYP2B6 induction potential and prediction of drug interactions via enzyme induction. Because all compounds in this study that enhanced CYP2B6 expression also induced CYP3A4, it was postulated that these enzymes share common regulation by hPXR and/or hCAR. This premise was supported further by similarities between the potencies and time course of CYP2B6 and CYP3A4 induction by select prototypical inducers. However, the relative contributions of hPXR and hCAR to CYP2B6 gene regulation remain unclear. Several lines of evidence support direct regulation of CYP2B6-inducible expression by hPXR. First, several studies have demonstrated that hPXR can bind to the NR1 and NR2 motifs present within the CYP2B6 PBREM (Xie et al., 2000; Goodwin et al., 2001; Mäkinen et al., 2002). Second, Goodwin et al. (2001) demonstrated that hPXR can mediate increased PBREM-driven reporter gene expression in response to rifampin. Third, a recent study showed that human PBREM was less selective for CAR compared with mouse PBREM, and that hPXR could compete with hCAR for binding to human PBREM (Mäkinen et al., 2002). Finally, a distal xenobiotic responsive enhancer module has been identified recently in the CYP2B6 promoter that binds hPXR and confers maximal responsiveness to CYP2B6 inducers in conjunction with the PBREM (Wang et al., 2003b). These previous findings are consistent with the similarities between the patterns of hPXR activation and CYP2B6 induction observed for most, but not all, inducers in this study.
In contrast to hPXR, no obvious correlation can be ascertained between the activation of hCAR and induction of CYP2B6. Despite the demonstrated role of CAR in the regulation of rodent CYP2B induction, it is uncertain whether hCAR plays an important role in CYP2B6 gene regulation because of species differences between rodent and human CAR. For example, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene can bind and activate mouse but not human CAR, and clotrimazole can deactivate hCAR but has a negligible effect on mouse CAR (Moore et al., 2000; Tzameli et al., 2000). Similarly, the androstane metabolites androstanol and androstenol repress the constitutive activity of mouse CAR but have relatively little effect on hCAR (Forman et al., 1998; Tzameli et al., 2000). Moreover, calcium/calmodulin-dependent kinase inhibitors disrupt PB induction of CYP2B expression and PBREM transactivation by rodent CAR but not hCAR (Zelko and Negishi, 2000).
In addition to pharmacological and biochemical differences between rodent and human CAR, the question of hCAR involvement in CYP2B6 regulation is complicated further by limitations of currently used cell-based transfection assays. Because CAR translocates spontaneously to the nucleus and exhibits high constitutive activity in transfected immortalized cells (Kawamoto et al., 1999), it is difficult to demonstrate convincingly the responsiveness of hCAR to CYP2B6 inducers. A compound such as androstanol that almost completely represses the constitutive activation of mouse CAR has not been identified for hCAR. Furthermore, it is uncertain whether measurement of CYP2B6 reporter gene activity in transfection assays is meaningful, given the possibility that hCAR may regulate CYP2B6 expression through mechanisms independent of ligand binding. Other mechanisms, such as nuclear translocation or interactions with other nuclear receptors and coactivator proteins, may dictate primarily the ability of hCAR to regulate CYP2B6 induction. For these reasons, the potential involvement of hCAR in CYP2B6 induction cannot be dismissed altogether, even though results from this study demonstrate that CYP2B6 reporter gene activities were not increased by any of the 14 compounds in hCAR-transfected cells. Alternative methods of evaluating hCAR are required before its relative contribution to CYP2B6 induction can be determined in a definitive manner.
Although CYP2B6 induction was closely correlated with CYP3A4 induction and hPXR activation for most inducing agents, some discrepancies were noted between the magnitudes of CYP2B6 and CYP3A4 induction in human hepatocytes, or between extents of hPXR activation in transfection assays and CYP2B6 induction in human hepatocytes. For example, CYP3A4 but not CYP2B6 was induced weakly by DEX. Since most studies have failed to demonstrate strong activation of PXR-mediated CYP3A4 reporter gene activity by DEX (Blumberg et al., 1998; Jones et al., 2000; Moore et al., 2000), a possible explanation for selective induction of CYP3A4 by DEX involves a PXR-independent mechanism as proposed by El-Sankary et al. (2002). These authors suggest that glucocorticoids regulate CYP3A4 gene expression through binding of activated GR to a nonconsensus glucocorticoid response element within the CYP3A4 promoter. Such an element may not be present within the CYP2B6 promoter, thus accounting for the lack of induction of this enzyme by DEX. Notably, basal CYP2B6 expression does not increase in primary hepatocytes treated with increasing concentrations of DEX between 1 and 100 nM, whereas that of CYP3A4 does (Pascussi et al., 2000; Wang et al., 2003a).
In contrast to DEX, CMZ, PHN, and PB demonstrated a greater magnitude of induction of CYP2B6 compared with CYP3A4. These compounds may act through other nuclear receptors, in addition to hPXR, that influence induction of CYP2B6 more than CYP3A4. For example, PB may induce CYP2B6 through both hPXR and hCAR pathways but induce CYP3A4 primarily through hPXR. Differential involvement of nuclear receptors in PB induction may account for the statistically significant difference observed between the EC50 values for CYP2B6 and CYP3A4. On the other hand, compounds such as PHN and CMZ may induce CYP2B6 and CYP3A4 via separate regulatory mechanisms. In CYP3A4 reporter gene assays, Luo et al. (2002) demonstrated approximately 3- and 8-fold activation of hPXR by CMZ and PHN, respectively. However, these inducers may act primarily through hCAR for CYP2B6, as suggested by their weak or negligible activation of hPXR in CYP2B6 reporter gene assays, but moderate to strong induction of CYP2B6 in human hepatocytes. The relative contributions of hPXR and hCAR to CYP2B6 induction are likely compound-specific and may be influenced by differences in the relative expression of each nuclear receptor, their affinities for response elements in CYP2B6 promoters, and their interactions with other regulatory proteins.
Finally, some discrepancies were noted between the magnitudes of hPXR activation of PBREM reporter gene expression and CYP2B6 induction. PAX was associated with strong hPXR activation but caused no discernible increases in CYP2B6 protein or activity in primary hepatocyte cultures. Its lack of effect on CYP2B6 protein or activity may stem from its cytotoxic effects in primary cultured cells due to inhibition of microtubule depolymerization. A previous study demonstrated lack of PB induction of CYP2B1/2 proteins in primary rat hepatocytes exposed to microtubule-disrupting agents such as PAX, suggesting that microtubule integrity is required for maintenance of signal transduction pathways leading to enzyme induction (Brown et al., 1995). A different explanation likely accounts for the disparities between moderate hPXR activation and weak CYP2B6 induction by DTBA and SPZ. One possibility is differences in sensitivity between the in vitro systems used to evaluate nuclear receptor activation and induction. Higher concentrations of DTBA and SPZ may be required to observe CYP2B6 induction in cultured primary hepatocytes than is required for reporter gene activation in transfection assays. Notably, DTBA and SPZ induced CYP3A4 expression by only 2- to 3-fold in primary human hepatocytes, similar to the degree observed for CYP2B6.
In conclusion, this report represents one of the most comprehensive investigations of CYP2B6 regulation because of the broad range of compounds examined with respect to concentration- and temporal-dependent inductive effects and nuclear receptor activation profiles. The results demonstrate that CYP2B6 is highly inducible by several known CYP3A4 inducers. Furthermore, the potencies and/or temporal kinetics of CYP2B6 and CYP3A4 induction were similar for CLZ, PB, PHN, and RIF, suggesting coordinate regulation of these enzymes by these inducers. The correspondence between the patterns of CYP2B6 induction and activation of hPXR for some inducers implicates this nuclear receptor in the regulation of CYP2B6-inducible expression. However, involvement of hCAR in this phenomenon cannot be excluded until other approaches are developed for assessing activation of hCAR by CYP2B6 inducers.
Acknowledgments
We thank Dr. John Cidlowski for provision of human GR expression vector and Dr. Gary Pollack for helpful suggestions on pharmacodynamic modeling and data analysis. Human liver tissue was procured with the assistance of Drs. Benjamin Calvo and Kevin Behrns, University of North Carolina at Chapel Hill Hospitals, and Lynn Johnson and Evageline Reynolds of the Lineberger Comprehensive Cancer Center Tissue Procurement Program, University of North Carolina.
Footnotes
-
↵1 Abbreviations used are: CAR, constitutive androstane receptor; BCIP/NBT, 5-bromo-4-chloroindolyl-phosphatase/nitrobenzotetrazolium; CLZ, clotrimazole; CMZ, carbamazepine; DEX, dexamethasone; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; DTBA, dexamethasone t-butylacetate; GR, glucocorticoid receptor; hCAR, human constitutive androstane receptor; hGRα, human glucocorticoid receptor α; hPXR, human pregnane X receptor; HL-N, human liver number; MTX, methotrexate; PAX, paclitaxel; PB, phenobarbital; PBREM, phenobarbital-responsive enhancer module; PHN, phenytoin; PROB, probenecid; PXR, pregnane X receptor; RIF, rifampin; RIT, ritonavir; SDM, sulfadimidine; SPZ, sulfinpyrazone; TAO, troleandomycin.
-
This work was supported in part by DuPont Pharmaceutical Company and by a grant from the National Institutes of Health (DK061652).
- Received April 2, 2003.
- Accepted December 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}