


Abstract
26,26,26,27,27,27-Hexafluoro-1α,25-dihydroxyvitamin D3 [F6-1α, 25(OH)2D3], which is now clinically used as a drug for secondary hyperparathyroidism, is a hexafluorinated analog of the active form of vitamin D3. Our previous studies demonstrated that CYP24A1 is responsible for the metabolism of F6-1α,25(OH)2D3 in the target tissues and that F6-1α,25(OH)2D3 was successively converted to F6-1α,23S,25(OH)3D3 and F6-23-oxo-1α,25(OH)2D3. In this study, we examined the metabolism of F6-1α,25(OH)2D3,F6-1α,23S,25(OH)3D3, and F6-23-oxo-1α,25(OH)2D3 by human UDP-glucuronosyltransferases (UGTs). Of these compounds, F6-1α,23S,25(OH)3D3 was remarkably glucuronidated both in human liver microsomes and in the recombinant system expressing human UGT. No significant interindividual differences were observed among 10 human liver samples. The recombinant system for 12 species of human UGTs revealed that F6-1α,23S,25(OH)3D3 glucuronidation was specifically catalyzed by UGT1A3. The information obtained in this study seems very useful to predict the metabolism and efficacy of vitamin D analogs in human bodies before clinical trials. In addition, note that for the first time a possible probe substrate for UGT1A3 has been found.
Vitamin D analogs are potentially useful for clinical treatments of type I rickets, osteoporosis, renal osteodystrophy, psoriasis, leukemia, and breast cancer (Bishop et al., 1994; Bouillon et al., 1995). 26,26,26,27,27,27-Hexafluoro-1α,25(OH)2D3, which is now clinically used as a drug for secondary hyperparathyroidism in cases of chronic renal failure and for the control of hypoparathyroidism (Nakatsuka et al., 1992; Akiba et al., 1998; Inoue and Fujimi, 1998; Mori et al., 1998), is a hexafluorinated analog of the active form of vitamin D3. F6-1α,25(OH)2D31 has been reported to be several times as potent as the parent compound at increasing intestinal calcium transport and bone calcium (Tanaka et al., 1984; Kiriyama et al., 1991; Inaba et al., 1993). The enhanced biological activity of F6-1α,25(OH)2D3 in such target tissues as kidneys and the small intestine is considered to be related to the conversion of F6-1α,25(OH)2D3 to F6-1α,23S,25(OH)3D3, a bioactive 23S-hydroxylated form resistant to further metabolism (Honda et al., 1993; Hayashi et al., 1998). Moreover, Komuro et al. (2003) demonstrated the local retention of [1β-3H]F6-1α,25(OH)2D3 and the bioactive metabolite F6-1α,23S,25(OH)3D3 in parathyroid glands after intravenous administration, indicating the higher potency of F6-1α,25(OH)2D3 than 1α,25(OH)2D3 in parathyroid glands. Our previous study demonstrated that CYP24A1 is responsible for the metabolism of F6-1α,25(OH)2D3 in the target tissues and that in recombinant Escherichia coli cells expressing human CYP24A1, bovine adrenodoxin, and NADPH-adrenodoxin reductase, F6-1α,25(OH)2D3 was successively converted to F6-1α,23S,25(OH)3D3 and F6-23-oxo-1α,25(OH)2D3 (Hayashi et al., 1998; Sakaki et al., 2003). In the liver, these metabolites were not detected due to the absence of CYP24A1. Instead, glucuronidation and 3-epimerization of F6-1α,25(OH)2D3 and F6-1α,23S,25(OH)3D3 were observed in rat livers as reported by Komuro et al. (1999).
The formation of glucuronides from such lipophilic substrates as steroids, bile acids, bilirubin, hormones, drugs, and environmental toxicants is catalyzed by UGTs, which have a large number of isozymes with different substrate specificities (Mackenzie et al., 1997). So far, 15 human UGT cDNAs have been identified, consisting of eight UGT1A proteins encoded in the UGT1A locus and seven proteins encoded in UGT2 genes (Tukey and Strassburg, 2000). The amino acid sequences within the UGT1A subfamily and those within the UGT2 family share 66 and 59% identity, respectively (Tukey and Strassburg, 2000). The distribution of individual isoforms leads to tissue-specific metabolism and detoxification. UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B10, 2B11, and 2B15 are expressed in the human liver (Tukey and Strassburg, 2000).
In this article, we demonstrate that F6-1α,23S,25(OH)3D3 is specifically glucuronidated by UGT1A3.
Materials and Methods
Materials. F6-1α,25(OH)2D3, F6-1α,23S,25(OH)3D3, and F6-23-oxo-1α,25(OH)2D3 were generous gifts from Sumitomo Pharmaceuticals (Osaka, Japan). UDP-glucuronic acid and β-glucuronidase were purchased from Sigma-Aldrich (St. Louis, MO).
Human liver microsomes from 10 individual human livers (HH13, HG112, HH47, HG95, HH18, HG43, HG74, HK25, HG89, and HG93) were purchased from BD Gentest (Woburn, MA). Recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10 UGT2B4, UGT2B7, UGT2B15, and UGT2B17 expressed in baculovirus-infected insect cells (Supersomes), and control microsomes from insect cells infected with wild-type baculovirus were also purchased from BD Gentest. All other chemicals were of the best commercially available grade.
Glucuronidation Assay. The reaction mixture contained 0.08 mg protein/ml human recombinant UGT microsomes or 0.1 mg protein/ml human liver microsomes, 20 mM MgCl2, 100 mM Tris-HCl, pH 7.4, 1 mM EDTA, and 2 mM UDP-glucuronic acid. Each of F6-1α,25(OH)2D3, F6-1α,23S,25(OH)3D3, and F6-23-oxo-1α,25(OH)2D3 in ethanol was added as a substrate at a final concentration of 1 to 20 μM. The reaction was initiated by the addition of UDP-glucuronic acid, and the reaction mixture was incubated at 37°C for 1 h.
Aliquots of the reaction mixture were extracted with 4 volumes of chloroform/methanol [3:1 (v/v)]. The organic phase was recovered and dried in a vacuum evaporator centrifuge (Sakuma Seisakusyo, Tokyo, Japan). The resultant residue was solubilized with the mixture of acetonitrile and water (1:1) and applied to HPLC under the following conditions: column, YMC-Pack ODS-AM [4.6 mm (inner diameter) × 300 mm] (YMC, Kyoto, Japan) UV detection, 265 nm; flow rate, 1.0 ml/min; column temperature, 40°C; and mobile phase, linear gradient of 20 to 100% acetonitrile aqueous solution containing 0.01% trifluoroacetic acid. On the other hand, an equal volume of acetonitrile was added to the aqueous phase, and the resultant solution was dried in a vacuum evaporator centrifuge without evaporating substrates and the metabolites. The resultant residue was analyzed by HPLC as described above.
Analysis of the Polar Metabolite of F6-1α,23S,25(OH)3D3. The metabolite observed in the aqueous phase was treated with β-glucuronidase. The putative glucuronide that was isolated from HPLC effluents was incubated for 1 h with 0.11 mg/ml β-glucuronidase in 20 mM potassium phosphate buffer, pH 7.4, at 37°C. The metabolite was analyzed as described above.
Kinetic Analysis. The kinetics studies were performed using human liver microsome (HH47) and recombinant human UGT1A3 expressed in baculovirus-infected insect cells (Supersomes). In determining the kinetic parameters, the F6-1α,23S,25(OH)3D3 concentration ranged from 0 to 30 μM. Kinetic parameters were determined on the basis of fitted curves using a computer program of KaleidaGraph (Synergy Software, Reading, PA). Although the kinetics of F6-1α,23S,25(OH)3D3 glucuronidation in recombinant UGT1A3 fitted to the Michaelis-Menten kinetics up to 3.5 μM F6-1α,23S,25(OH)3D3, the glucuronidation activity gradually decreased with increasing substrate concentration at more than 3.5 μM. Therefore, the following equations were applied for Michaelis-Menten kinetics (eq. 1) using the activities at 0 to 3.5 μM or substrate inhibition kinetics (eq. 2) using the activities at 0 to 30 μM. On the other hand, the kinetics of F6-1α,23S,25(OH)3D3 glucuronidation in human liver microsomes fitted to the Michaelis-Menten kinetics at the full range of substrate concentrations. The equation was applied for Michaelis-Menten kinetics (eq. 1), 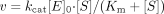

where [S] and [E]0 (nanomoles per milligram of protein) are the substrate concentration and UGT contents, respectively. The Km, Ki, and kcat represent a Michaelis-Menten constant, a substrate inhibition constant (Houston and Kenworthy, 2000), and a catalytic rate constant, respectively.
Kinetic parameters of F6-1α,23S,25(OH)3D3 glucuronidation by human liver microsomes (HH47) were determined from eq. 3 based on the Michaelis-Menten two-enzyme model: 
LC-MS Analysis of the Metabolites. Isolated metabolites from HPLC effluents were subjected to mass spectrometric analysis using a Finnegan Mat TSQ-70 with atmospheric pressure chemical ionization, positive mode. The conditions of LC were described below: column; reverse phase ODS column (6 × 150 mm) (μBondapak C18; Waters, Milford, MA); mobile phase, 80% methanol aqueous solution; flow rate, 1.0 ml/min; and UV detection, 265 nm.
Results
F6-1α,23S,25(OH)3D3 Glucuronidation by Recombinant UGT Isoforms. Of the three substrates examined, human liver microsomes showed a remarkable activity toward F6-1α,23S,25(OH)3D3, whereas no glucuronides of F6-1α,25(OH)2D3 and F6-23-oxo-1α,25(OH)2D3 were detected in recombinant UGT isoforms and human liver microsomes (data not shown). Thus, we further examined metabolism of F6-1α,23S,25(OH)3D3 by 12 recombinant UGT isoforms expressed in baculovirus-infected insect cells. Figure 1 shows the HPLC profile of the metabolite (M1) and substrate [F6-1α,23S,25(OH)3D3] recovered from the aqueous phase and the organic phase, respectively. The retention time of the metabolite of F6-1α,23S,25(OH)3D3 formed in some of the recombinant UGT isoforms was 17.4 min.
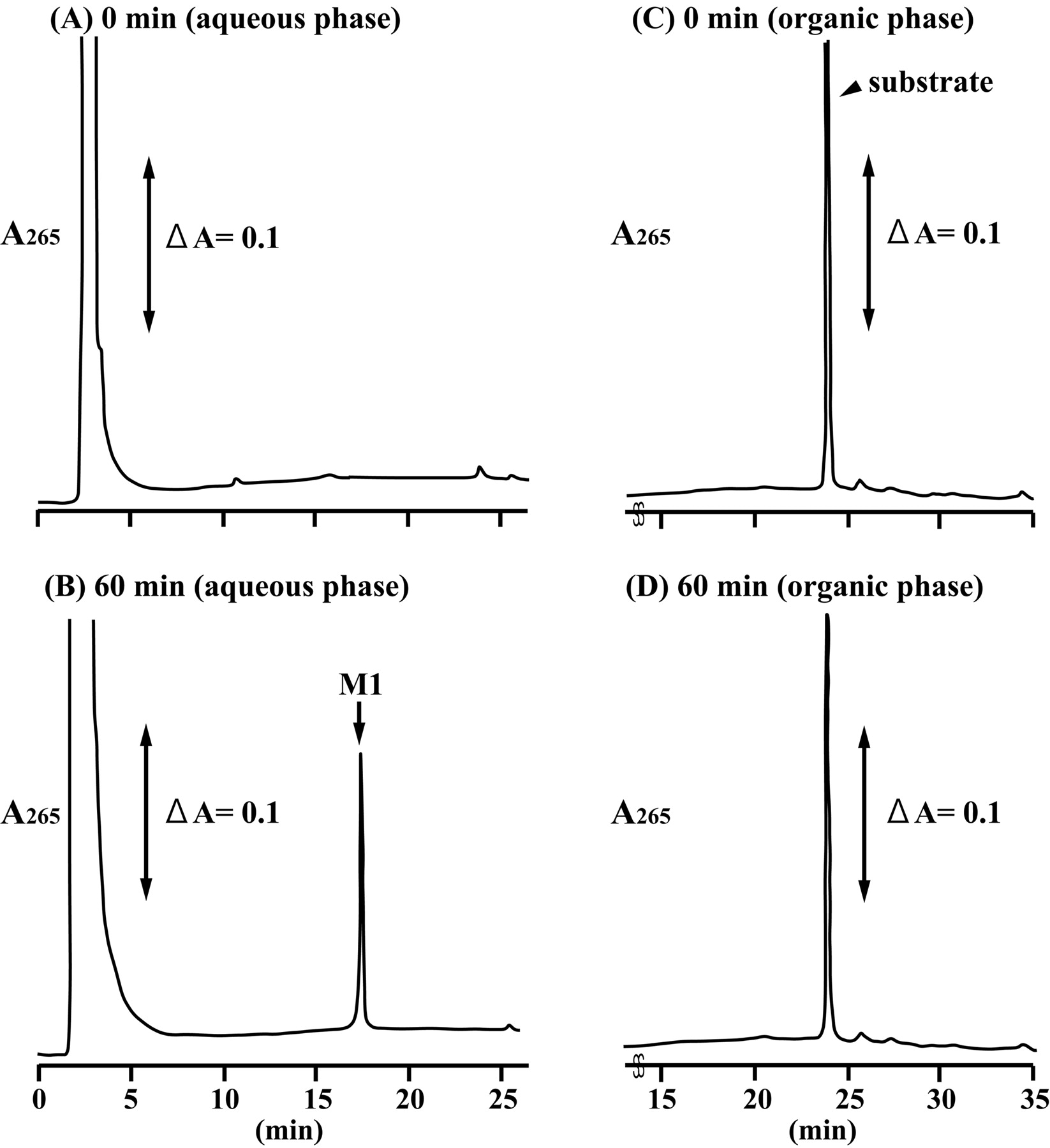
HPLC profiles of F6-1α,23S,25(OH)3D3 and its metabolite by the recombinant human UGT1A3. The organic phase and the aqueous phase at the reaction time of 0 min (A and C) and 60 min (B and D) were analyzed by HPLC as described under Materials and Methods. The elution position of the metabolite (M1) was indicated.
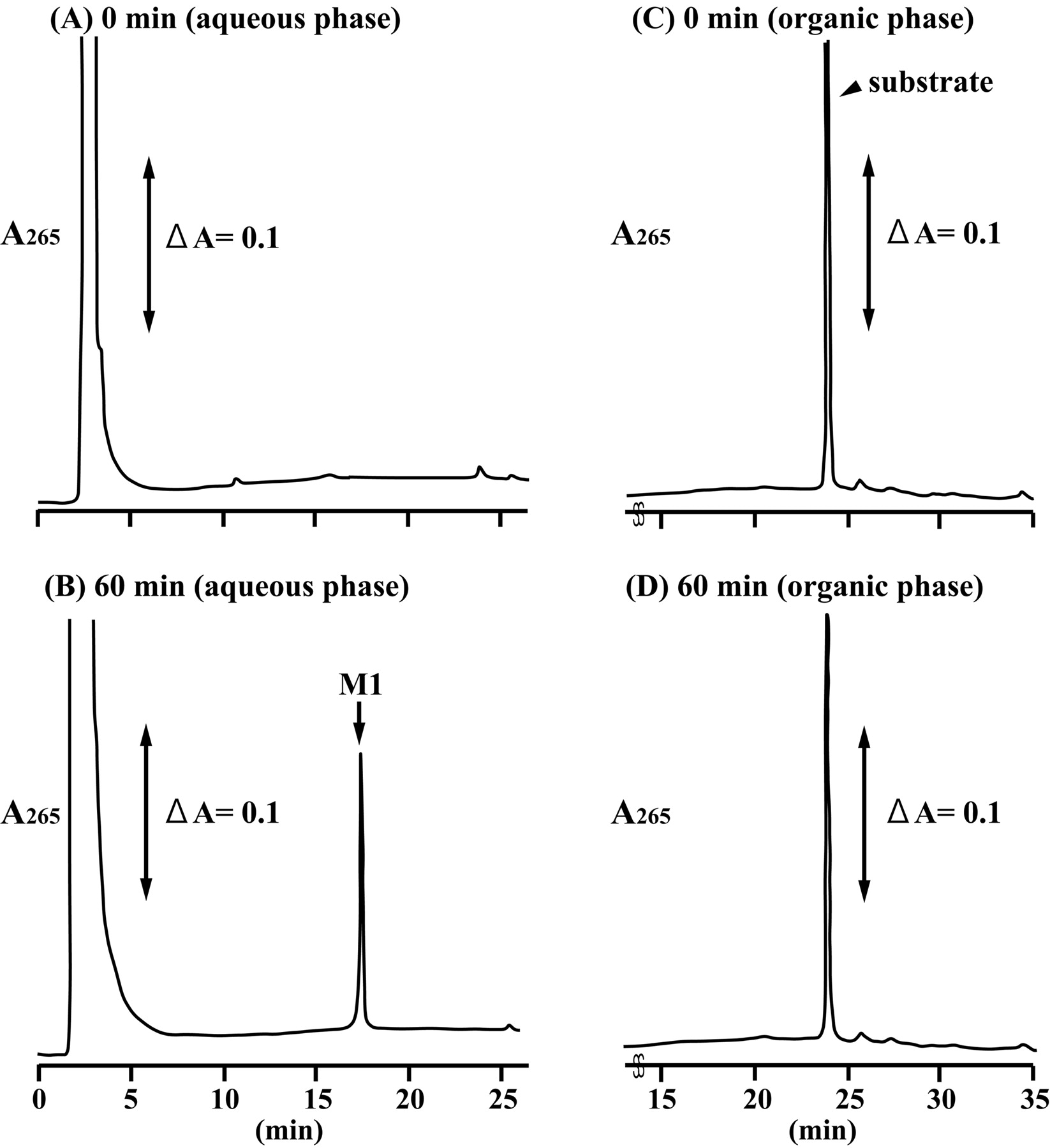
The glucuronidation activities were measured at the substrate concentration of 1, 5, and 20 μM because recombinant UGT1A3 showed substrate inhibition kinetics as described below. The contents of UGT1A1, 1A3, 1A4,1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15, and 2B17 in the microsomes were estimated to be 1.7, 0.8, 1.4, 1.6, 1.0, 1.1, 1.0, 1.6, 0.3, 0.6, 0.3, and 0.6 nmol/mg protein, respectively, as described previously (Kasai et al., 2004). Based on the contents of UGTs, molecular-based activity (v/[E]0) was calculated. As shown in Fig. 2, UGT1A3 showed a remarkable glucuronidation activity toward F6-1α,23S,25(OH)3D3. Although UGT1A4, UGT2B4, and UGT2B7 also showed the activity, their activities were significantly low compared with that of UGT1A3.

F6-1α,23S,25(OH)3D3 glucuronidation activity in the recombinant UGT isoforms expressed in baculovirus-infected insect cells. F6-1α,23S,25(OH)3D3 glucuronidation activity in the recombinant UGT microsomes was examined at 1 μM (white column), 5 μM (dotted column), and 20 μM (black column) F6-1α,23S,25(OH)3D3. N.D., not detected.
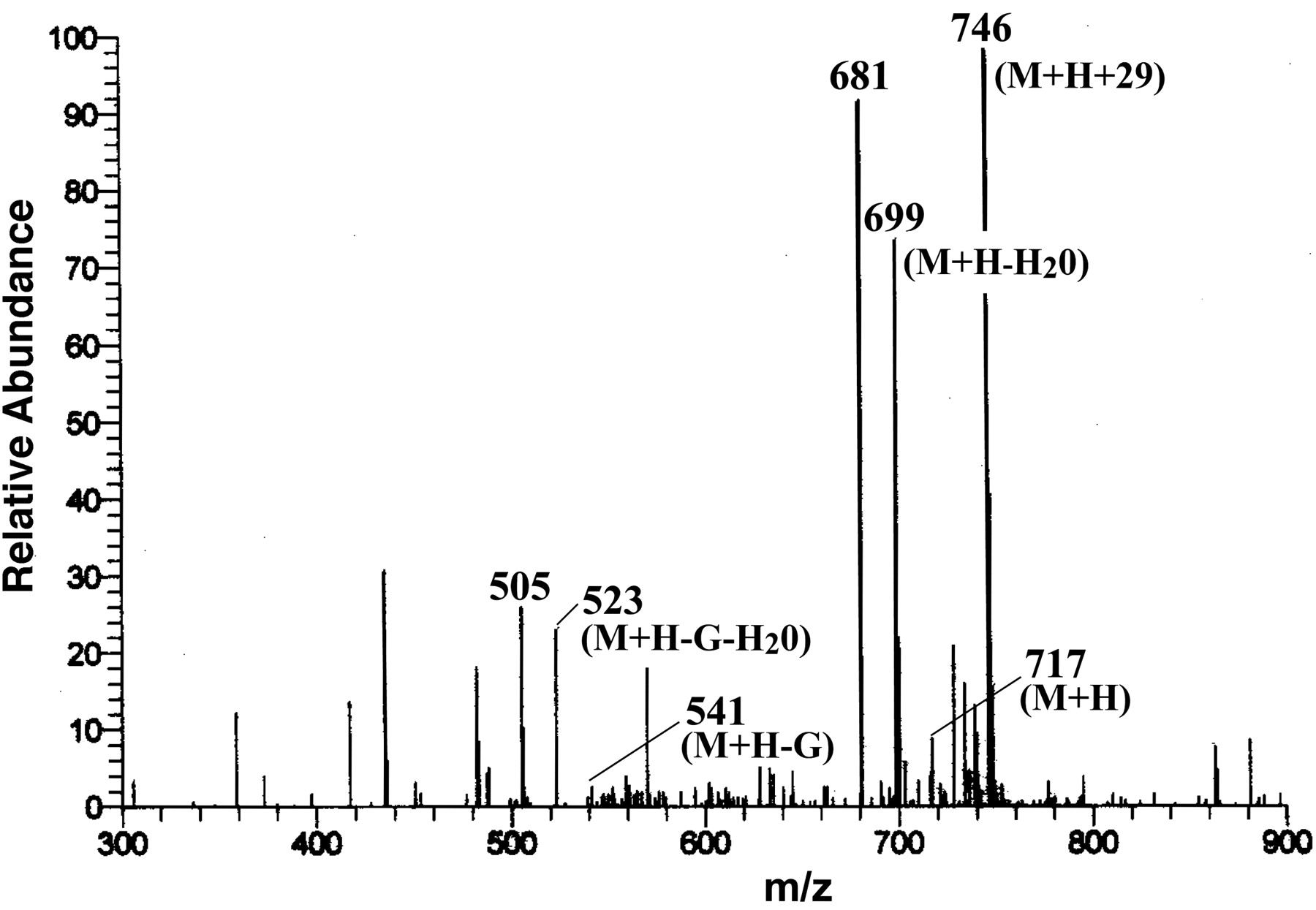
LC-MS Analysis of the Metabolite (M1). The polar metabolite (M1) of F6-1α,23S,25(OH)3D3 was converted to F6-1α,23S,25(OH)3D3 by β-glucuronidase, suggesting that M1 is F6-1α,23S,25(OH)3D3 glucuronide (data not shown). We also analyzed M1 by LC-MS to confirm that M1 is F6-1α,23S,25(OH)3D3 glucuronide. As shown in Fig. 3, the metabolite M1 showed a molecular ion at m/z 717 (M + H), and fragment ions at 699 (717-H2O), 688 (717-2H2O), 541 (717-G), 523 (717-G-H2O), and 505 (717-G-2H2O). These results strongly suggest that the metabolite is O-glucuronide of F6-1α,23S,25(OH)3D3. The ion at m/z 746 (M + H + 29), which has been characteristically observed under the LC-MS conditions (Sawada et al., 2000), was also observed.

Mass spectrum of the metabolite M1 shown in Fig. 1. G, glucuronide.
F6-1α,23S,25(OH)3D3 Glucuronidation Activity in the Microsomes from 10 Human Livers. The retention time of the metabolite of F6-1α,23S,25(OH)3D3 formed in human liver microsomes was identical to that of F6-1α,23S,25(OH)3D3 formed in the recombinant human UGTs, suggesting that the metabolite was the glucuronide of F6-1α,23S,25(OH)3D3. F6-1α,23S,25(OH)3D3 glucuronidation activities in microsomes from 10 human livers were examined at 5 μM F6-1α,23S,25(OH)3D3 (Fig. 4). The activity range was 2.4-fold.

F6-1α,23S,25(OH)3D3 glucuronidation activity in the microsomes from 10 human livers. The activity was measured at 5 μM F6-1α,23S,25(OH)3D3 as described under Materials and Methods.
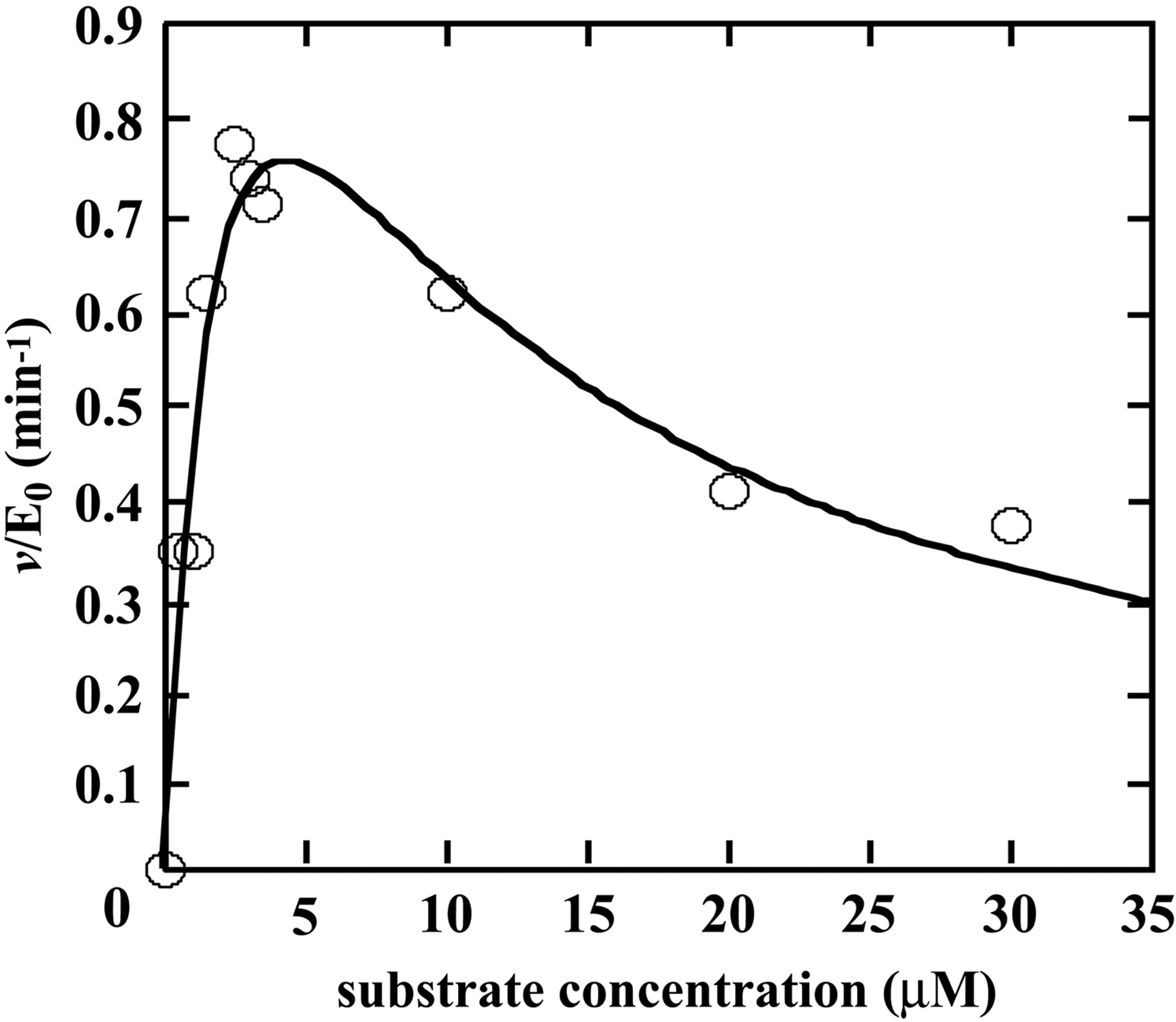
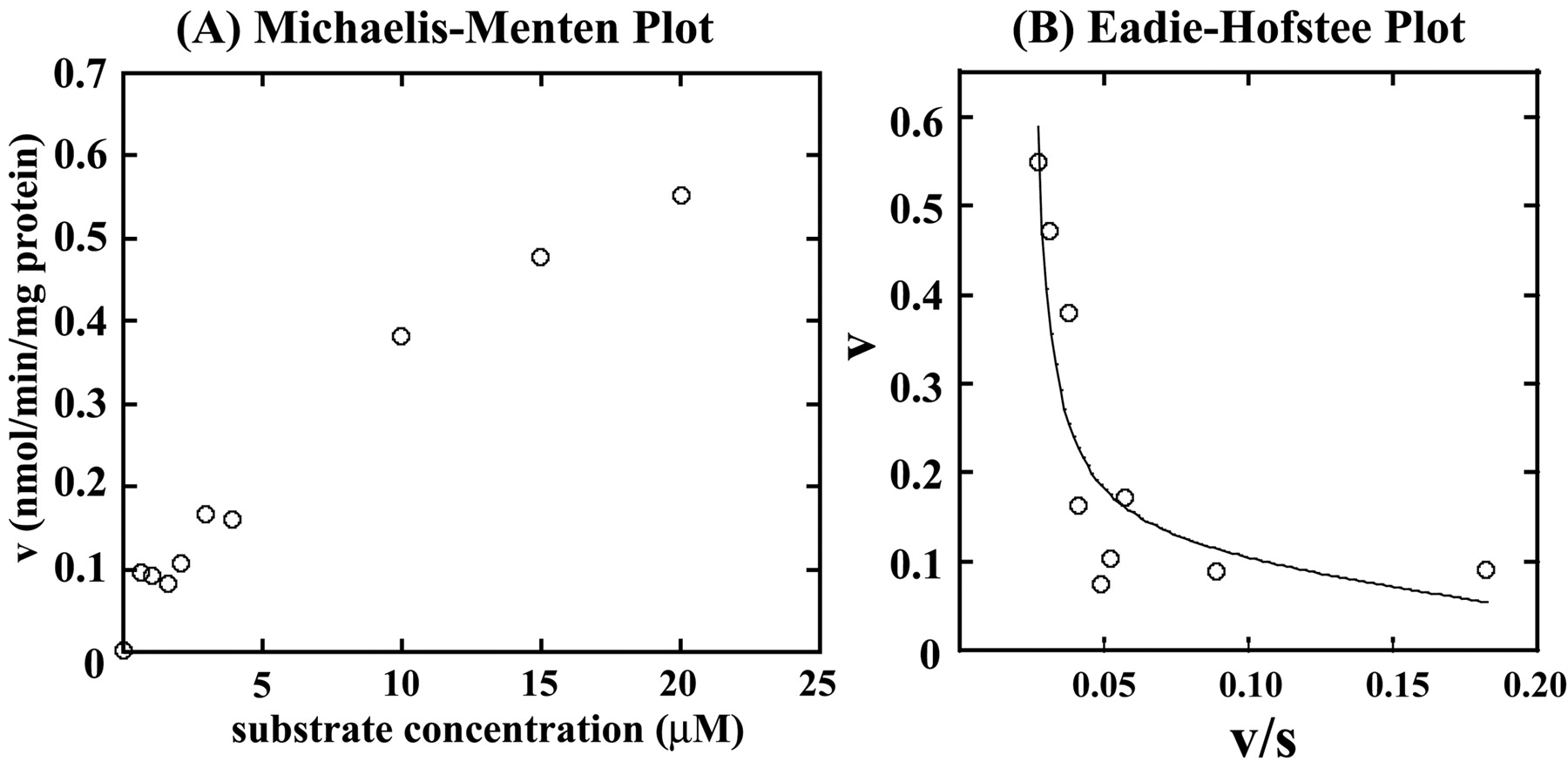
Kinetic Analysis of F6-1α,23S,25(OH)3D3 Glucuronidation by Recombinant Human UGT1A3. As shown in Fig. 2, F6-1α,23S,25(OH)3D3 glucuronidation seems to be specifically catalyzed by UGT1A3. Therefore, we examined kinetics of UGT1A3 (Fig. 5). When kinetic parameters were determined by fitting to the eq. 1 using initial velocity values at 0 to 3.5 μM F6-1α,23S,25(OH)3D3, the apparent kinetic parameters, Km and kcat, were estimated to be 0.92 ± 0.47 μM and 0.78 ± 0.25 min-1, respectively. On the other hand, kinetic parameters, Km, kcat, and Ki, determined by fitting to the eq. 2 at 0 to 30 μM F6-1α,23S,25(OH)3D3, were estimated to be 2.24 ± 0.07 μM, 1.35 ± 0.29 min-1, and 6.17 ± 2.70 μM, respectively. Kinetic Analysis of F6-1α,23S,25(OH)3D3 Glucuronidation by Human Liver Microsomes (HH47). The kinetics studies were performed using human liver microsomes (HH47). In determining the kinetic parameters, the F6-1α,23S,25(OH)3D3 concentration ranged from 0 to 20 μM (Fig. 6). As shown in Fig. 6A, human liver microsomes (HH47) did not show substrate inhibition kinetics. Other human liver microsomes did not show substrate inhibition kinetics either (data not shown). Because human liver microsomes contain multiple forms of UGT, the kinetic parameter Vmax (nanomoles per minute per milligram of protein) was determined. The apparent kinetic parameters Km and Vmax were estimated to be 16.17 ± 1.89 μM and 1.09 ± 0.10 nmol/min/mg protein from eq. 1, respectively. As shown in Fig. 6B, however, the Eadie-Hofstee plots showed biphasic, suggesting that multiple UGT isoforms are involved in glucuronidation of F6-1α,23S,25(OH)3D3 in human liver microsomes. Thus, the kinetic parameters were also determined by fitting to the Michaelis-Menten two-enzyme model. For the high-affinity component, Km1 and Vmax1 were 0.41 μM and 0.11 nmol/min/mg protein, respectively, whereas for the low-affinity component, Km2 and Vmax2 were 2420 μM and 54 nmol/min/mg protein, respectively. It is noted that the Vmax1/Km1 value is 12-fold higher than the Vmax2/Km2 value.

Kinetic analysis of F6-1α,23S,25(OH)3D3 glucuronidation by recombinant human UGT1A3. The dots fitted to the eq. 2. Kinetic parameters were determined as described under Materials and Methods.

Kinetic analysis of F6-1α,23S,25(OH)3D3 glucuronidation in the human liver microsome (HH47). Michaelis-Menten plots (A) and Eadie-Hofstee plots (B). The dots fitted to eq. 3, and kinetic parameters were determined as described under Materials and Methods.
Discussion
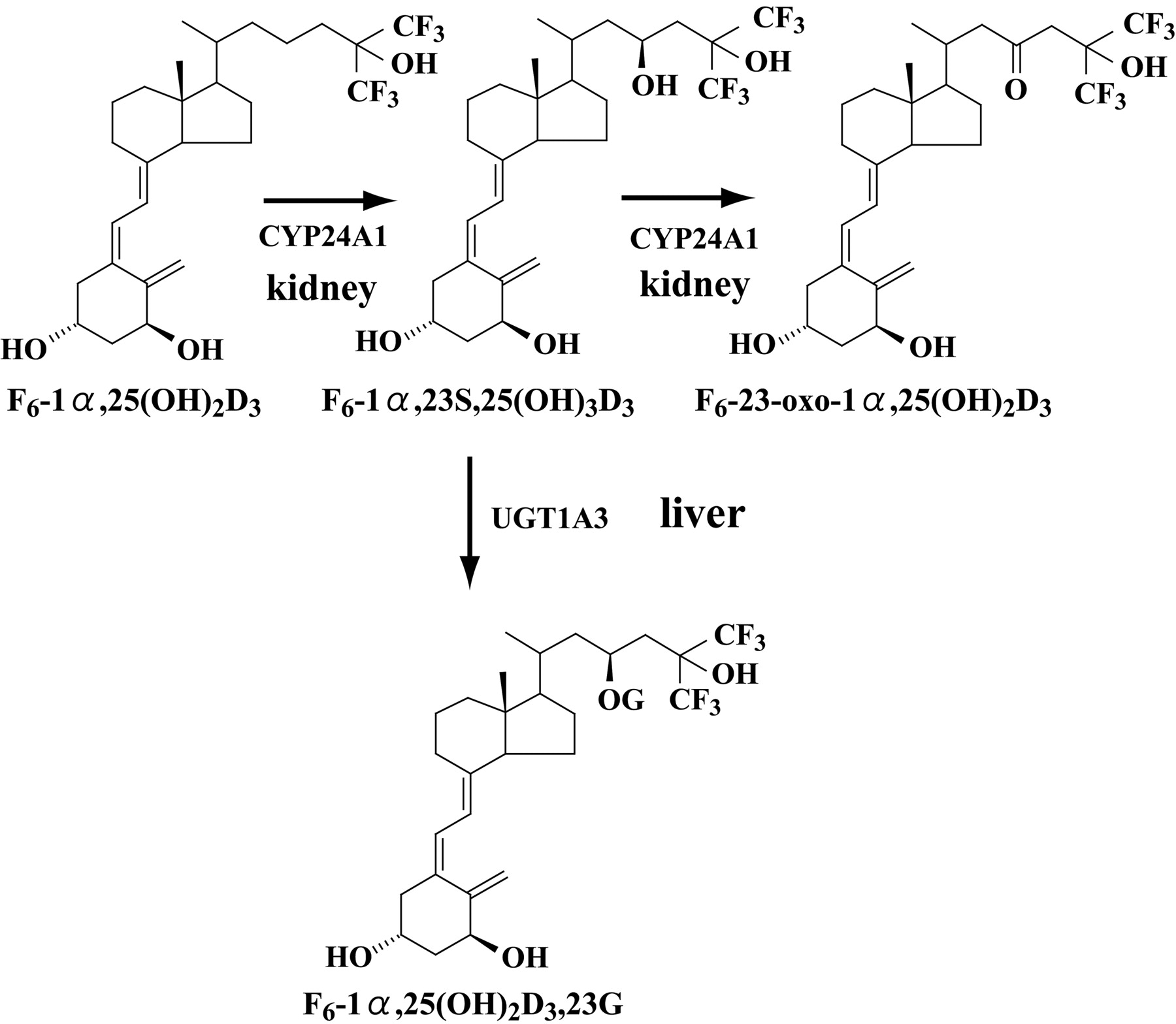
Our previous studies demonstrated that human CYP24A1 was responsible for the conversion of F6-1α,25(OH)2D3 to inactive metabolite via F6-1α,23S,25(OH)3D3 and F6-23-oxo-1α,25(OH)2D3 and that the slow conversion from F6-1α,23S,25(OH)3D3 to F6-23-oxo-1α,25(OH)2D3 caused the accumulation of F6-1α,23S,25(OH)3D3 (Sakaki et al., 2003). In this study, it was demonstrated that F6-1α,23S,25(OH)3D3 was glucuronidated in recombinant UGT isoforms and human livers microsomes. The glucuronide of F6-1α,23S,25(OH)3D3 was observed, whereas no glucuronides of F6-1α,25(OH)2D3 and F6-23-oxo-1α,25(OH)2D3 were detected, suggesting that the hydroxyl group at C-23 position is more readily glucuronidated than the other three hydroxyl groups at C-1α, C-3β, and C-25 positions. Furthermore, Fig. 2 suggests that human UGT1A3, which exists in human liver (Mojarrabi et al., 1996), specifically contributes to the glucuronidation of F6-1α,23S,25(OH)3D3. Human UGT1A3 is a well known principal enzyme for the metabolism of many therapeutic agents that have aglycones that are aryl, primary, secondary, tertiary aliphatic, or heterocyclic to acyl-O-glucuronides (carboxylic esters) (Mojarrabi et al., 1996; Cheng et al., 1998; Green et al., 1998; Green and Tephly, 1998; Strassburg et al., 1999; Hiller et al., 1999; Nowel et al., 1999). Based on our previous work and this study, the major metabolic pathway of F6-1α,25(OH)2D3 in human bodies seems to be 23-hydroxylation by CYP24A1 in the kidney followed by glucuronidation by UGT1A3 in the liver (Fig. 7). As described previously (Sakaki et al., 2003), F6-1α,23S,25(OH)3D3 and F6-23-oxo-1α,25(OH)2D3 still have a high affinity for vitamin D receptor. Therefore, glucuronidation of F6-1α,23S,25(OH)3D3 seems to be an important step to decrease the affinity for vitamin D receptor. The kinetics of UGT1A3-dependent glucuronidation of F6-1α,23 25(OH)3D3 showed substrate inhibition as shown in Fig. 5. Substrate inhibition phenomena also can be seen in the kinetics of UGT1A3-dependent glucuronidation of estrone (Iwai et al., 2004) and troglitazone glucuronidation by UGT1A1 and UGT1A10 (Watanabe et al., 2002). Note that kinetics of F6-1α,23S,25(OH)3D3 glucuronidation in human liver microsomes did not show the substrate inhibition. As shown in Fig. 6B, the Eadie-Hofstee plots showed biphasic kinetics. Thus, kinetic parameters were determined by fitting to the Michaelis-Menten two-enzyme model. For the high-affinity component, kinetic parameter Km was estimated to be 0.41 μM. Although the activity of UGT1A4 is much lower than the activity of UGT1A3, UGT1A4 exhibited a similar pattern to UGT1A3 as shown in Fig. 2. On the other hand, UGT2B4 and UGT2B7 seems to have low affinity for F6-1α,23S,25(OH)3D3 judging from the activity at substrate concentrations of 1, 5, and 20 μM (Fig. 2). Thus, it might be possible to assume that the low-affinity components in the human liver are UGT2B4 and UGT2B7, although their contents in human liver are unknown. In the clinical treatment by F6-1α,25(OH)2D3, however, F6-1α,23S,25(OH)3D3 concentration in human bodies may not be high enough to cause substrate inhibition. Therefore, UGT1A3 seems to be responsible for F6-1α,23S,25(OH)3D3 glucuronidation in human liver. As shown in Fig. 4, 2.4-fold interindividual differences were observed, which seem to be derived from the different expression level of UGT1A3 in the human livers or UGT1A3 polymorphisms reported by Iwai et al. (2004). Based on these results, it is possible to assume that F6-1α,23S,25(OH)3D3 may be a probe substrate for human UGT1A3. Several probe substrates for each UGT have already been found: for example, bilirubin for UGT1A1 and propofol for UGT1A9. Probe substrates for such UGT isoforms as UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A10, and UGT2B15, however, have not been clearly identified (Tukey and Strassburg, 2000). It is interesting that a great difference was observed in the F6-1α,23S,25(OH)3D3 glucuronidation between UGT1A3 and UGT1A4, despite sharing 93% of their amino acid sequence (Tukey and Strassburg, 2000). Although their different substrate specificities have often been reported so far, which amino acid residue is responsible for the difference remains unsolved. Thus, genetic engineering studies of UGT1A3 and UGT1A4 may increase understanding of the structure-function relationship of UGTs.

Sequential metabolism of F6-1α,25(OH)2D3 by CYP24A1 in the kidney and by UGT1A3 in the liver in human bodies.
Acknowledgments
We express our gratitude to Dr. S. Komuro (Sumitomo Chemical Co. Ltd.) for useful discussions.
Footnotes
-
This work was partly supported by Health Science research grants from the Ministry of Health, Labor and Welfare of Japan, and a grant-in-aid for scientific research from the Ministry of Education, Science, Sports and Culture of Japan.
-
doi:10.1124/dmd.104.002303.
-
ABBREVIATIONS: F6-1α,25(OH)2D3, 26,26,26,27,27,27,-hexafluoro-1α,25-dihydroxyvitamin D3; UGT, UDP-glucuronosyltransferase; HPLC, high-performance liquid chromatography; LC-MS, liquid chromatography-mass spectrometry.
- Received September 14, 2004.
- Accepted October 20, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}