


Abstract
Mycophenolic acid (MPA), the active metabolite of the immunosuppressant mycophenolate mofetil is primarily metabolized by glucuronidation. The nature of UDP-glucuronosyltransferases (UGTs) involved in this pathway is still debated. The present study aimed at identifying unambiguously the UGT isoforms involved in the production of MPA-phenyl-glucuronide (MPAG) and MPA-acylglucuronide (AcMPAG). A liquid chromatography-tandem mass spectrometry method allowing the identification and determination of the metabolites of mycophenolic acid was developed. The metabolites were characterized in urine and plasma samples from renal transplant patients under mycophenolate mofetil therapy and in vitro after incubation of mycophenolic acid with human liver (HLM), kidney (HKM), or intestinal microsomes (HIM). The UGT isoforms involved in MPAG or AcMPAG production were investigated using induced rat liver microsomes, heterologously expressed UGT (Supersomes), and chemical-selective inhibition of HLM, HKM, and HIM. The three microsomal preparations produced MPAG, AcMPAG, and two mycophenolate glucosides. Among the 10 UGT isoforms tested, UGT 1A9 was the most efficient for MPAG synthesis with a Km of 0.16 mM, close to that observed for HLM (0.18 mM). According to the chemical inhibition experiments, UGT 1A9 is apparently responsible for 55%, 75%, and 50% of MPAG production by the liver, kidney, and intestinal mucosa, respectively. Although UGT 2B7 was the only isoform producing AcMPAG in a significant amount, the selective inhibitor azidothymidine only moderately reduced this production (approximately -25%). In conclusion, UGT 1A9 and 2B7 were clearly identified as the main UGT isoforms involved in mycophenolic acid glucuronidation, presumably due to their high hepatic and renal expression.
The prodrug mycophenolate mofetil is an immunosuppressive agent widely used for the prevention or treatment of acute rejection after solid organ transplantation. Mycophenolate mofetil is given orally, absorbed and converted to mycophenolic acid, the active immunosuppressant form. Mycophenolic acid (MPA) acts by inhibition of type II inosine monophosphate dehydrogenase, a key enzyme in the de novo purine biosynthetic pathway. Mycophenolic acid is mainly metabolized by glucuronidation to its inactive hydroxy-β-glucuronide (MPAG) (Bullingham et al., 1998). MPAG is partly excreted into the bile and contributes to mycophenolic acid enterohepatic circulation after deglucuronidation in the gut (Bullingham et al., 1998). In addition, two other metabolites, i.e., MPA-acyl-glucuronide (AcMPAG) and MPA-phenyl-glucoside (MPAGls), were isolated in renal transplant patients' plasma and identified by tandem mass spectrometry (Schütz et al., 1999; Shipkova et al., 1999). It was reported that AcMPAG had an inhibitory effect on lymphocyte proliferation in vitro (Shipkova et al., 2001b). On the other hand, the toxicity of acyl-glucuronides is well known (Ritter, 2000; Shipkova et al., 2003): they can bind covalently to proteins and other macromolecules, which is thought to be the mechanism of their immunogenicity and toxicity. AcMPAG might thus be responsible for some of the adverse effects of mycophenolate mofetil therapy, such as leukopenia or gastrointestinal toxicity (Wieland et al., 2000). MPA glucosides (MPAGls) did not exhibit immunosuppressive activity (Schütz et al., 1999), and nothing is known about their toxicity.
Significant pharmacokinetic interactions between mycophenolate mofetil and other immunosuppressants have been described. Mycophenolic acid exposure is lower when cyclosporine is associated with mycophenolate mofetil, which would be due to inhibition of the enterohepatic cycling of mycophenolic acid, although further evidence of this relationship is needed (Smak Gregoor et al., 1999). Mycophenolic acid in vivo concentrations and in vitro immunosuppressant activity seem to be higher when mycophenolate mofetil is administered with tacrolimus than with cyclosporine, with a corresponding reduction of MPAG concentrations (Zucker et al., 1997; Hübner et al., 1999). This suggests an inhibition of mycophenolic acid metabolism by tacrolimus, further demonstrated in vitro (Zucker et al., 1999). Steroids also interfere with mycophenolic acid bioavailability, likely through induction of the UDP-glucuronosyltransferase (UGT) isoforms involved in MPAG formation, as described by Cattaneo et al. (2002). No data regarding possible drug-drug interactions resulting in concentration variations of the active and presumably toxic AcMPAG are available. A better knowledge of the metabolic pathways of mycophenolic acid is necessary to better investigate such interactions.
To date, at least 15 forms of UGTs have been identified in humans. Based on sequence similarities, they have been divided into two families, UGT 1 and UGT 2. The UGT isoforms involved in the glucuronidation of mycophenolic acid to MPAG were previously studied by several teams, but remain debated. Morissette et al. (2001) demonstrated that mycophenolic acid glucuronidation into MPAG is more intensive in men than in women, suggesting that mycophenolic acid could compete with estrogens for the UGT 1A class. Shipkova et al. (2001) suggested that most of the UGT 1A and 2B were involved in MPA metabolism, with UGT 1A7 and 2B4 being the most active. For Mackenzie (2000), UGT 1A8, 1A9, and 1A10 have the capacity to glucuronidate mycophenolic acid, whereas UGT 1A1, 1A3, 1A4, 1A6, 2B4, 2B7, and 2B10 are inactive (Mojarrabi and Mackenzie, 1997). Basu et al. (2004) and Bernard and Guillemette (2004) also found that UGT 1A7, 1A8, 1A9, and 1A10 were able to produce MPAG, but there were substantial differences in the catalytic activities observed: for Bernard and Guillemette (2004), UGT 1A8 and 1A9 are the main isoforms producing MPAG and UGT 1A1, 1A7, and 1A10 have low capacities, whereas Basu et al. (2004) found catalytic activities as follows: UGT 1A7 > 1A9 > 1A8 > 1A10. Finally, it is worth noting that the UGT isoforms producing AcMPAG have only been studied by Basu et al. (2004) using nonspecific methods (i.e., hydrolysis of AcMPAG and determination of mycophenolic acid using thin-layer chromatography).
This study describes the full in vitro/in vivo characterization of the different phase II metabolites of mycophenolic acid, some of which have been identified by only one team so far (Shipkova et al., 1999), and the investigation of the UGT isoforms responsible for the formation of MPAG and, above all, AcMPAG, using complementary in vitro models and a specific and sensitive LC-MS/MS method.
Materials and Methods
Chemicals and Reagents. Mycophenolic acid, indomethacin, UDP-glucose, UDP-glucuronic acid, type 2 β-glucuronidase (EC 3.2.1.31), β-glucosidase (EC 3.2.1.21), Triton X-100, 3-methylcholanthrene, phenobarbital, and propofol were obtained from Sigma-Aldrich (St. Louis, MO), azidothymidine from GlaxoSmithKline (Nanterre, France), MgCl2 from Merck (Darmstadt, Germany), and MPAG from Analytical Services International Ltd. (London, UK). All reagents were analytical grade.
In Vivo Investigation of Mycophenolic Acid Metabolites. Mycophenolic acid metabolites were investigated in urine and plasma samples collected from renal transplant recipients (n = 14), grafted for more than 30 days, who had given their informed consent to participate in a pharmacokinetic study of cyclosporine and mycophenolate mofetil. All patients received 1 g of mycophenolate mofetil twice daily and a dose of cyclosporine adjusted to yield a trough level of 150 to 200 ng/ml. Urine and plasma samples were acidified with 10 μl/ml phosphoric acid to prevent AcMPAG degradation (Shipkova et al., 2000) and then stored at -20°C until analysis. The interdose areas under the concentration-time curve (AUC0-12h) of mycophenolic acid, AcMPAG, MPAG, and MPAGls were calculated using the linear trapezoidal rule, and the average metabolic ratio of each metabolite was calculated in each patient.
In Vitro Metabolic Experiments.Human Microsomes. Pooled human liver microsomes were obtained from BD Gentest (Woburn, MA, USA). Pooled human kidney and intestinal microsomes were prepared as previously described (Picard et al., 2004) from normal kidney (n = 4) and ileal (n = 4) tissues adjoining tumor tissues from surgical specimens, with the approval of the local ethics committee. Protein concentration of the microsomal suspension was measured according to the Bradford method (Bradford, 1976) using bovine serum albumin as standard.
Rat Liver Microsomes. Male Sprague-Dawley rats were treated with phenobarbital sodium and 3-methylcholanthrene. Phenobarbital, dissolved in 0.9% NaCl, was administered i.p. at a dose of 70 mg/kg once daily for 3 days; 10 mg/kg per day of 3-methylcholanthrene, in solution in corn oil, were injected i.p. for 3 days. NaCl without phenobarbital or corn oil without 3-methylcholanthrene was administered following the same protocol to control rats. The rats were then killed by decapitation and liver was removed. Rat liver microsomes were obtained by differential centrifugation as previously described (Picard et al., 2004).
Recombinant UGTs. Microsomes prepared from baculovirus-infected insect cells that expressed the human UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, and 2B7 (Supersomes), as well as a control preparation, were obtained from BD Gentest.
The glucuronidation activity of Supersomes from each UGT were substantiated by the supplier for the following substrates: estradiol (UGT 1A1 and 1A3), trifluoperazine (UGT 1A4), and 7-hydroxy-4-trifluoromethylcoumarin (UGT 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, and 2B7).
Incubation Procedures. The typical incubation mixture (500 μl) contained 0.5 mg/ml microsomal proteins (HLM, HKM, HIM, rat liver microsomes, or Supersomes), 2 mM UDP-glucuronic acid or UDP-glucose, 10 mM MgCl2, 0.025 to 1 mM mycophenolic acid, and 0.1 M Tris-HCl buffer (pH 7.4). Microsomes were detergent-activated by preincubation with Triton X-100 during 30 min on ice, with a detergent-to-microsomal protein ratio of 0.4 (w/w). Mycophenolic acid and microsomes were preincubated at 37°C for 5 min before starting the reaction by addition of the cosubstrate. After 30 min of incubation at 37°C, the reaction was stopped by addition of 25 μl of perchloric acid (24%, v/v). After centrifugation, the supernatant was stored at -20°C until analysis. Control incubations without microsomes or without cosubstrates were performed in parallel. After checking for the linearity of metabolite formation with increasing microsomal protein concentration (0.1-1 mg/ml) and incubation time (10-60 min), kinetic determinations were performed using a microsomal protein concentration of 0.5 mg/ml (HLM, HKM, HIM, or Supersomes) and a 30-min incubation time.
Chemical Inhibition Experiments. The individual contribution of UGT 1A9 and 2B7 in MPAG or AcMPAG production was evaluated in HLM, HKM, and HIM using propofol (Ebner and Burchell, 1993) and azidothymidine (Barbier et al., 2000; Court et al., 2003) as selective substrates, respectively. The inhibitors were tested at 4 increasing concentrations (50-500 μM) to assess the maximal selective inhibition. The same incubation procedure as described above was followed, except that inhibitors were added to the incubation medium before starting the reaction. Inhibitors were dissolved in dimethyl sulfoxide and control incubations were spiked with the same amount of dimethyl sulfoxide (≤1%, v/v). MPAG or AcMPAG formation rates in the presence of the inhibitors were compared with those of the corresponding controls and the percentage of inhibition was calculated.
Hydrolysis Experiments. To confirm the position of the glucuronide and glucoside moieties on MPA, the effect of alkaline or enzymatic hydrolysis on metabolites produced in vitro was tested.
Reaction mixtures (1 ml) containing the target glucuronides or glucosides were generated by incubating 0.5 mM mycophenolic acid and 0.5 mg/ml microsomes as described above. Then, 200 μl of each incubation medium were further incubated during 1 h at 37°C in the presence or absence (control) of either β-glucuronidase (100 units) or β-glucosidase (20 units) in 50 μl of 1 M sodium acetate buffer (pH 4.8). In parallel, 200 μl of the same supernatants were submitted to alkaline hydrolysis by incubation with or without 0.5 M NaOH (10 μl) during 2 h at room temperature. The concentration of conjugates was determined using LC-MS/MS, and the hydrolysis rate was calculated.
LC-MS/MS Analysis.Identification of Mycophenolic Acid Metabolites. Identification experiments were performed after solid-phase extraction. Incubation medium samples (500 μl) diluted 1/3 with water were passed through a 60-mg Oasis MAX extraction cartridge (Waters, Milford, MA) previously activated with 2 ml of methanol followed by 2 ml of H2O. The cartridge was rinsed with 2 ml of 50 mM, pH 7 sodium acetate/methanol (95:5, v/v), and elution was achieved with 1 ml of methanol containing 2% formic acid. The extract was evaporated to dryness and reconstituted in 50 μl of acetonitrile/ammonium formate (2 mM, pH 3.0) (30:70, v/v). The chromatographic system consisted of two series 200 micro-flow rate, high-pressure gradient pumps (PerkinElmer Life and Analytical Sciences, Boston, MA) and an Innertsil ODS-3 C18, 5-μm (150 × 2.1 mm i.d.) reversed-phase column (GL Sciences, Tokyo, Japan). The mobile phase, delivered at a constant flow rate of 200 μl/min, was a gradient of solution A [acetonitrile (2 mM, pH 3.0)/ammonium formate (90:10, v/v)] in solution B (2 mM, pH 3.0, ammonium formate), as follows: 0 to 2 min, 20% A; 10 min, 50% A; 12 min, 90% A; 12 to 15 min, 90% A; 20 min, 20% A. The detection was performed using a QTRAP (Applied Biosystems/MDS Sciex, Foster City, CA) tandem mass spectrometer with linear ion-trapping capabilities in the third quadrupole. A Turbo-Ionspray source was used in the negative ionization mode with a spray voltage of -4500 V. Identification of mycophenolic acid metabolites was achieved using the following steps: acquisitions were first performed in the precursor ion scan mode to find metabolites giving rise to a fragment of the same mass as mycophenolic acid molecular ion (m/z 319), i.e., phase II metabolites. The nature of the metabolites detected was then confirmed using the enhanced product ion (EPI) scan mode, offering a high sensitivity by means of linear ion-trapping. Then, three-stage mass spectrometry (MS3) experiments were used to investigate the fragmentation pathways and metabolite structure.
Determination of Mycophenolic Acid and Metabolites. Mycophenolic acid and MPAG in renal transplants were determined using a previously described, fully validated LC-MS/MS method (Prémaud et al., 2004). AcMPAG, MPAGls, and AcMPAGls were determined using the same method with minor modifications: specific transitions of mycophenolic acid (m/z 319→191; 319→287), mycophenolic acid glucuronides (m/z 495→319; 495→175), mycophenolic acid glucosides (m/z 481→319; 481→162), and indomethacin as internal standard (m/z 356→112) were monitored in the multiple reaction monitoring (MRM) mode. MPA and MPAG quality controls in plasma prepared in-house were analyzed with each run. AcMPAG, AcMPAGls, and MPAGls concentrations were estimated as MPAG molar equivalents with respect to MPAG calibration curves because of the lack of the respective pure compounds. The minor AcMPAGls was determined in vitro but not in renal transplants due to very low concentrations. The limit of quantification of the assay was 10 ng/ml for MPAG. Linearity was verified up to 1500 ng/ml (r = 0.999). The between-day precision CVs and mean relative errors were less than 10% over the linearity range.
Kinetic Modeling. All in vitro data points represent the mean of duplicate determination. Microsomal kinetic data were model-fitted and apparent Km and Vmax calculated according to the Michaelis-Menten model using Winreg 3.1 software (available online: http://www.unilim.fr/pages_perso/jean.debord/winreg/winreg1.htm).
Results
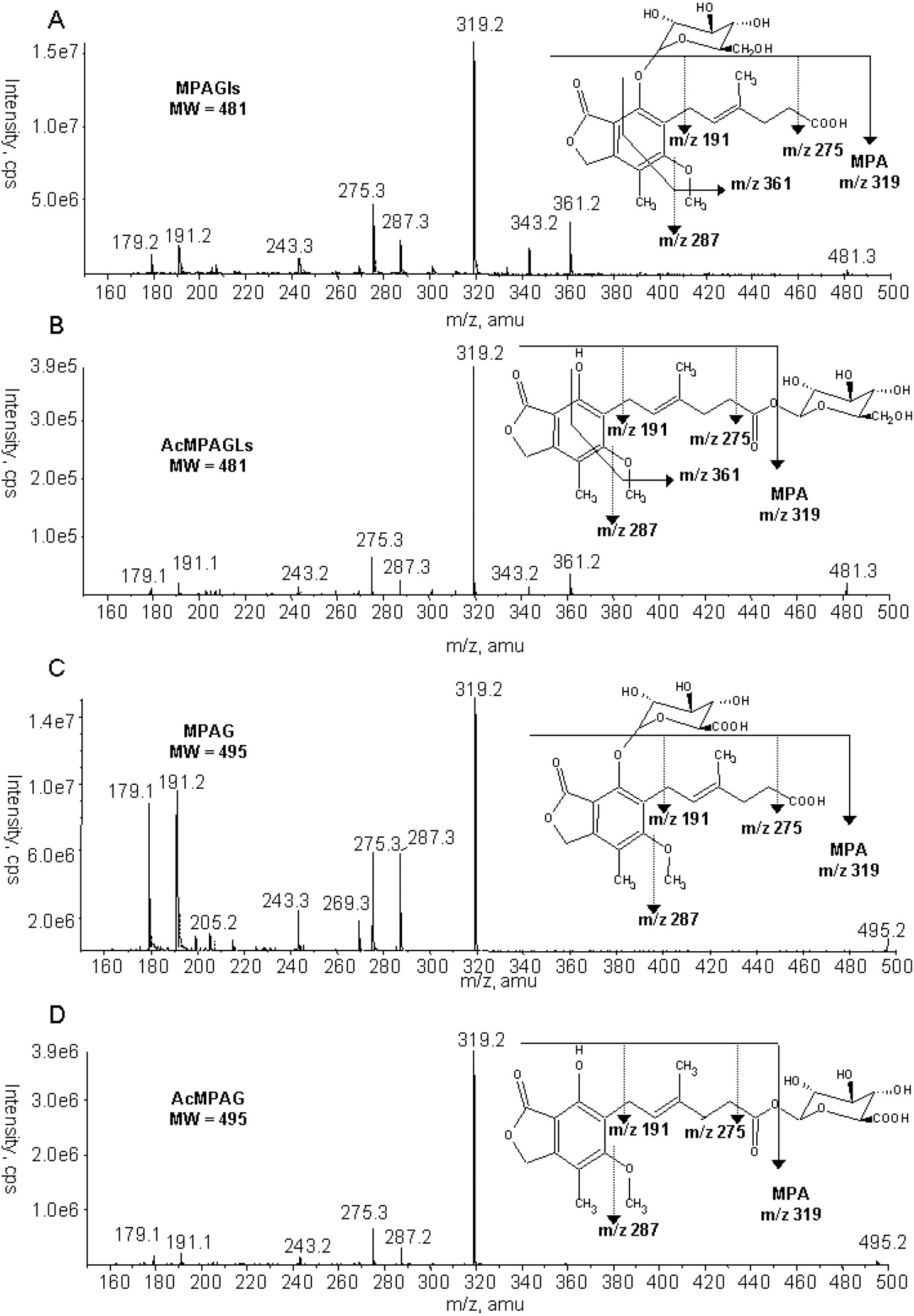
In addition to MPAG, three potential metabolites of mycophenolic acid were detected in urine samples using the precursor ion scan mode, where compounds producing [mycophenolic acid] as major fragment ion were monitored (i.e., mycophenolic acid phase II metabolites). Analysis in the MRM mode confirmed their nature (Fig. 1): two glucuronides (MPAG and AcMPAG) were detected by following the specific MRM transitions (m/z 495→319; m/z 495→175). The other two metabolites were found with MRM transitions corresponding to the loss of a glucosyl moiety (m/z 481→319; m/z 481→162), confirming the existence of two mycophenolic acid glucosides. By comparison with the glucuronides, the first-eluted and major glucoside was supposed to be MPAGls and the second and minor glucoside, AcMPAGls. The full mass spectra of the metabolites were obtained in the EPI scan mode (Fig. 2): fragmentation of all the metabolites led to mycophenolic acid (m/z 319) and secondarily to mycophenolic acid fragment ions as confirmed by MS3 acquisitions. The two glucosides showed a specific fragment ion (m/z 361), but no fragment could give hints about the conjugation position (phenolic or acyl group). This was solved out by hydrolysis experiments which confirmed that MPAG (RT = 10.0 min) and the supposed MPAGls (RT = 8.6 min) were stable to alkaline hydrolysis and subject to complete degradation by β-glucuronidase and β-glucosidase, respectively, whereas AcMPAG (RT = 13.6 min) and the supposed AcMPAGls (RT = 9.4 min) were degraded under alkaline conditions and stable to enzymatic hydrolysis (Table 1), as previously reported for AcMPAG (Shipkova et al., 1999).
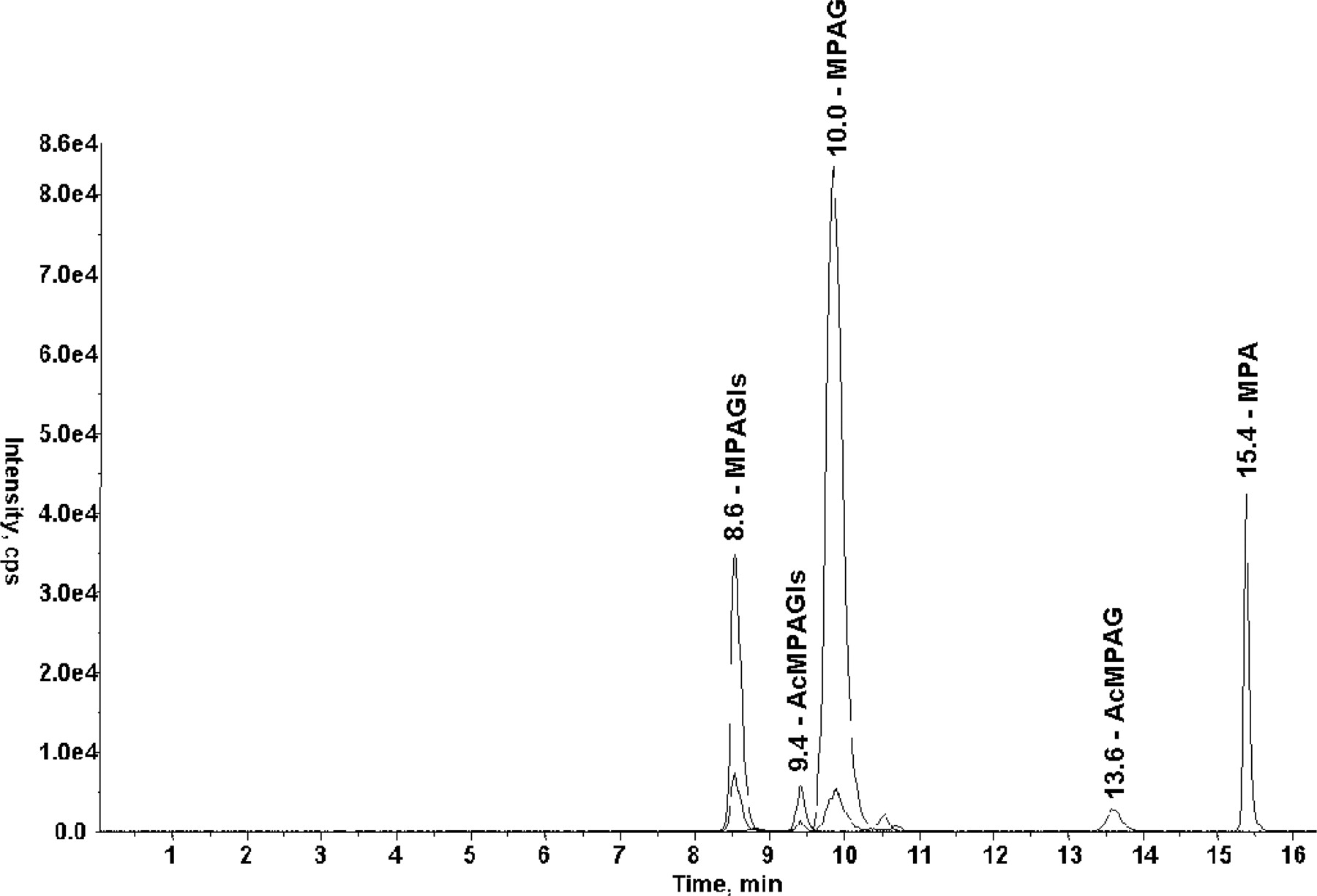
Chromatograms of the m/z 319→191 (mycophenolic acid), m/z 495→319 (mycophenolic acid glucuronides), and m/z 481→319 (mycophenolic acid glucosides) MRM transitions obtained from a urine sample of a renal transplant recipient under mycophenolate mofetil and cyclosporine therapy.

MS/MS spectra (EPI mode) of mycophenolic acid metabolites produced in vitro by human liver microsomes. MW, molecular weight.
Alkaline and enzymatic hydrolysis of the glucuronide and glucoside conjugates of mycophenolic acid produced in vitro
Results are presented as the mean percentage of hydrolysis with respect to controls (duplicate experiments).
Additionally, purified fractions corresponding to the metabolites' retention times (MPAG, AcMPAG, and MPAGls) were obtained by a modification of the LC-MS/MS method to a preparative mode, then assayed using NMR (data not shown). The structure of MPAG was confirmed, but the signal/noise ratio was insufficient to confirm the putative AcMPAG and MPAGls.
In renal transplants, the average MPAG AUC0-12h was 11.2 to 37.2 times higher (average 24.1 times) than the mycophenolic acid average AUC0-12h. Apparent AcMPAG AUC0-12h represented 2.9 to 11.8% (mean = 6.2%) of mycophenolic acid AUC0-12h. Apparent MPAGls AUC0-12h ranged from 1.7 to 11.1% (mean = 5.4%) of mycophenolic acid AUC0-12h.
In Vitro Metabolism of Mycophenolic Acid. When mycophenolic acid was incubated with HLM, HKM, or HIM in the presence of UDP-glucuronic acid, the two glucuronides previously identified (MPAG and AcMPAG) were produced, as confirmed by retention time and MS/MS spectra. Similarly, incubation with UDP-glucose led to the formation of the two glucosides identified in biological fluids. The microsomal kinetic parameters of MPAG, AcMPAG, and MPAGls formation were calculated for HLM, HKM, and HIM (Table 2). Those of AcMPAGls were calculated for HLM and HKM but not for HIM because of a low activity. The maximal formation rate (Vmax) of MPAG was about 2.5 times and Km 3 times higher for kidney than for liver, resulting in mycophenolic acid intrinsic clearance (CLint: Vmax/Km ratio) almost similar to that of both types of microsomes. The apparent Km for AcMPAG formation was similar for kidney and liver, whereas Vmax was 3 times higher for liver, leading to a CLint 3 times as much for liver as for kidney microsomes. According to CLint values, mycophenolic acid-phenyl-glucosylation was the second main elimination pathway for mycophenolic acid. HLM or HKM produced MPAGls with the same efficiency, whereas HLM was more efficient than HKM for AcMPAGls production with much greater affinity. HIM showed the lowest activity of the three microsomal preparations for all the metabolites produced (Table 2).
Kinetic constants for glucuronide and glucoside formation by pooled human liver (HLM), kidney (HKM), and intestinal microsomes (HIM)
Results are means of duplicate measurements.
Mycophenolic Acid Glucuronidation by Induced Rat Liver Microsomes. Rat liver microsomes incubated with mycophenolic acid in the presence of UDP-glucuronic acid produced two glucuronides with the same retention times and MS/MS spectra as MPAG and AcMPAG, respectively. With microsomes obtained from 3-methylcholanthrene-pretreated rat liver, MPAG formation was enhanced by 1.9-fold, whereas there was no effect on AcMPAG production. The pretreatment of rats with phenobarbital enhanced AcMPAG (1.5-fold) but not MPAG synthesis (Table 3).
Effect of drug induction of UGTs on MPAG and AcMPAG formation by rat liver microsomes
Results are means of duplicate measurements.
Mycophenolic Acid Glucuronidation by Human Recombinant UGT. Among the 10 UGT isoforms tested, UGT 1A9, 1A8, 1A7, and 1A10 produced MPAG in significant amounts, with UGT 1A9 being the most active. UGT 1A1, 1A6, 2B7, and 2B4 showed little activity. AcMPAG production was mainly detected with UGT 2B7 and, to a much lesser extent, with UGT 1A1 and 2B4. No metabolite production was detected with UGT 1A3, 1A4, and control Supersomes (Fig. 3).

MPAG and AcMPAG apparent formation rates measured using incubation experiments with recombinant human UGT isoforms (Supersomes) at 0.1 mM mycophenolic acid concentration.
The kinetic parameters of MPAG and AcMPAG production were determined for the competent UGT isoforms (Table 4), except UGT 2B4, 1A6, and 1A10, whose metabolite formation rate was so low that kinetic parameters could not be calculated. UGT 1A9 showed a Km of 0.16 mM for MPAG formation, close to the Km calculated for HLM (0.18 mM) and higher than UGT 1A7 and 1A8 affinity. UGT 2B7 was the isoform most efficiently producing AcMPAG.
Kinetic parameters of MPAG and AcMPAG formation derived from incubations with recombinant human UGT isoforms (Supersomes)
Chemical Inhibition of Mycophenolic Acid Glucuronidation. The UGT 1A9 probe propofol decreased MPAG formation in a concentration-dependent manner up to 47.0% for HIM, 55.3% for HLM, and 76.3% for HKM relative to the respective controls. In contrast, there was no significant effect on AcMPAG formation by HLM, HKM, or HIM. Inhibition of AcMPAG formation by the UGT 2B7-selective substrate azidothymidine was quite constant for all the concentrations tested, with a maximum of 24.6% for HLM and 26.1% for HKM. In contrast, no inhibition was found for HIM. Azidothymidine inhibition of MPAG production by HLM, HKM, or HIM was less than 10% (Fig. 4).

Effect of increasing concentrations of two specific UGT chemical inhibitors (propofol/UGT 1A9 and azidothymidine/UGT 2B7) on MPAG and AcMPAG production by human (HLM; A), kidney (HKM; B), and intestinal (HIM; C) microsomes. Results are expressed as the mean percentage of inhibition with respect to controls.
Discussion
The existence and nature of two glucuronides of mycophenolic acid, namely, MPAG and AcMPAG, and two glucosides, as previously described by Shipkova et al. (1999), were confirmed in vivo and in vitro using LC-MS/MS. Based on hydrolysis experiments, the second glucoside found in minor amount was identified as the acylglucoside of mycophenolic acid; this metabolite was previously observed in vitro, after incubation of mycophenolic acid with human renal microsomes, but not in vivo (Shipkova et al., 2001a). The monitoring of specific ion transitions (MRM mode) allowed the specific and sensitive determination of these metabolites in vitro and in vivo.
Several clinical studies highlighted the fact that the kidney may contribute to mycophenolic acid metabolism based on comparison of urinary clearance of MPAG after oral or intravenous administration of mycophenolate mofetil and on the impact of hepatic impairment on MPAG clearance (Bullingham et al., 1996; Parker et al., 1996). In vitro studies provided additional evidence: Zucker et al. (1999) reported that purified kidney extracts contained higher amounts of UGT involved in MPAG formation than did liver extracts; comparison of kidney and liver microsomes for MPAG formation showed a higher activity of the former (Bowalgaha and Miners, 2001; Shipkova et al., 2001a; Bernard and Guillemette, 2004). Accordingly, we confirmed that MPAG maximal formation rate (Vmax) was 3 times higher for HKM than for HLM, with kinetic parameters quite similar to those previously reported (Vietri et al., 2000; Bowalgaha and Miners, 2001; Shipkova et al., 2001a; Bernard and Guillemette, 2004). Our results also suggest that AcMPAG is mainly produced by the liver, which is in accordance with the results of Shipkova et al. (2001a). Mycophenolic acid glucosides seem to be produced by both liver and kidney; but, contrary to Shipkova et al. (2001a), who reported that the acylglucoside was produced by the kidney, the best affinity was observed here with liver microsomes, with 7.5 times higher efficiency than kidney microsomes. In any case, the low maximal formation rate in vitro and concentrations in vivo confirmed that AcMPAGls is a minor metabolite of mycophenolic acid.
Contradictory data on the UGT isoforms involved in MPAG production and their respective contribution were available, and only one recent report studied the isoforms producing AcMPAG, using UGT-transfected cell lines and thin-layer chromatography (Basu et al., 2004). In the present study, experiments using induced rat liver microsomes clearly showed that different UGT isoforms were involved in MPAG and AcMPAG formation. Phenobarbital and 3-methylcholanthrene are two inducers of the cytochrome P450 and UGT isoforms. Phenobarbital (inducer of the rat UGT 1A1 and 2B1) enhanced the formation of AcMPAG, and it is worth noting that UGT 2B7 is described as the human ortholog of the rat UGT 2B1 (Ritter, 2000), these two isoforms having many substrates in common. Moreover, the rat UGT 2B1 and the human UGT 2B7 appear to be major contributors to glucuronidation of carboxylic acid drugs to acylglucuronides (Ritter, 2000). In contrast, MPAG formation was enhanced by 3-methylcholanthrene, which is a reported inducer of the rat and human hepatic UGT 1A6 (Grams et al., 2000). The UGT isoforms involved in mycophenolic acid glucuronidation were further identified by incubation with recombinant enzymes (UGT Supersomes). UGT 2B7 was confirmed to be responsible for production of AcMPAG, with higher activity in the liver. This UGT is expressed in liver, kidney, and small bowel as demonstrated by immunoblot analysis and enzymatic assays (Czernik et al., 2000; Fisher et al., 2001; Antonio et al., 2003). When mycophenolic acid was coincubated with the selective 2B7 substrate azidothymidine, only a slight inhibition of AcMPAG production was observed with HLM and HKM and none with HIM. Thus, we cannot rule out that other, unexplored UGT isoforms could contribute to AcMPAG production. Nevertheless, this result could be explained by the low affinity of azidothymidine (HLM Km = 1.4 mM; Court et al., 2003) as compared with mycophenolic acid (Km = 0.37 mM). The other known UGT2B7 probes (i.e., morphine, codeine) have lower specificity and higher Km, and thus lower inhibitory potency. The UGT isoforms involved in MPAG formation have been previously studied using recombinant UGT cell lysates (Shipkova et al., 2001a): all the isoforms tested were active, with UGT 1A7 and 2B4 being the most efficient. UGT 1A7 is only located in the small bowel, and the use of a specific antibody directed against human UGT 2B4 demonstrated that this isoform was exclusively expressed in the liver (Pillot et al., 1993). On the other hand, Mackenzie (2000) reported that the intestinal UGT 1A8 and 1A10 and the hepatic and renal UGT 1A9 were involved in MPAG formation using microsomes prepared from UGT-transfected cells and thin-layer chromatography. Finally, the recent reports of Basu et al. (2004) and Bernard and Guillemette (2004) described UGT 1A7, 1A8, 1A9, and 1A10 as the major contributors to MPAG production. According to both incubation with human recombinant UGT isoforms and chemical inhibition experiments, the present report clearly shows that UGT 1A9 is the main isoform involved, with at least 55% contribution to the hepatic MPAG production. UGT 1A1 and 1A6 probably account for a part of MPAG hepatic production, whereas UGT 1A7, 1A8, and 1A10 which are located in the small bowel, could contribute to mycophenolic acid intestinal first-pass effect. The recent work by Basu et al. (2004) using in situ hybridation of UGT1A9 mRNA showed that this isoform was also located in duodenal and ileal mucosa and thus could partly contribute to mycophenolic acid intestinal metabolism. Our results, based on chemical inhibition of UGT 1A9 by propofol, suggest that this isoform may be involved for approximately 40% of MPAG intestinal production. Nevertheless, as previously reported (Bowalgaha and Miners, 2001; Shipkova et al., 2001a), HIM showed much lower activity than kidney or liver microsomes, which is presumably due to a lower expression of UGT 1A9 in the intestine wall. Although UGT 1A9 was originally described as a liver-expressed UGT, the important contribution of the kidney to mycophenolic acid metabolism and the 75% decrease in MPAG production by kidney microsomes after selective UGT 1A9 inhibition are consistent with many recent reports about its higher expression in the kidney: McGurk et al. (1998) found that the glucuronidation of propofol, a specific substrate of UGT 1A9, was higher in human kidney than in liver. UGT 1A9 mRNA levels were also found to be higher in human kidney than in liver (Albert et al., 1999). Thus, the high kidney MPAG formation rate found in this study as well as in previous reports (Zucker et al., 1999; Vietri et al., 2000; Shipkova et al., 2001a; Bernard and Guillemette, 2004) is apparently mainly due to UGT 1A9.
Glucuronidation is an important feature of mycophenolic acid pharmacokinetics. Although the main metabolite MPAG is inactive, its AUC0-12h is on average more than 20 times higher than that of mycophenolic acid. There is no doubt that this high metabolic rate contributes significantly to mycophenolic acid elimination and enterohepatic cycling. Thus, glucuronidation could partly explain the high interindividual variability of mycophenolic acid AUC0-12h for a fixed 1-g b.i.d. dose. The confirmation and (for AcMPAG) identification of the UGT isoforms involved in mycophenolic acid glucuronidation presented here will allow new investigations on mycophenolic acid pharmacogenetic-pharmacokinetic relationships, drug-drug interaction, and adverse events. UGT 2B7, which is responsible for the production of the toxic acyl-glucuronide metabolite, is muted in approximately 50% of the white population, and this polymorphism could lead to a difference in metabolite production and, thus, in the hematological or digestive susceptibility to mycophenolic acid.
Acknowledgments
We gratefully thank Dr. J. C. Szelag for providing us with patients' samples.
Footnotes
-
This study was funded by the Limousin Regional Council and Limoges University Hospital.
-
doi:10.1124/dmd.104.001651.
-
ABBREVIATIONS: MPA, mycophenolic acid; MPAG, mycophenolic acid glucuronide; AcMPAG, mycophenolic acid acyl-glucuronide; MPAGls, mycophenolic acid glucoside; AcMPAGls, mycophenolic acid acyl-glucoside; MRM, multiple reaction monitoring; EPI, enhanced product ion; HLM, human liver microsomes; HKM, human kidney microsomes; HIM, human intestinal microsomes; UGT, UDP-glucuronosyltransferase; LC-MS/MS, liquid chromatography-mass spectrometry; AUC0-12h, interdose areas under the concentration-time curve; RT, retention time; CLint, intrinsic clearance.
- Received July 28, 2004.
- Accepted October 4, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}