


Abstract
From fits of drug transport kinetics across confluent MDCKII-hMDR1-NKI and Caco-2 cell monolayers we estimated the levels of efflux active P-glycoprotein (P-gp) in these two cell lines (companion paper). In the present work, we compared the efflux active P-gp number to the total P-gp level, using liquid chromatography–tandem mass spectrometry, and showed that in Caco-2 cells total P-gp is about 10-fold greater than efflux active P-gp, whereas in MDCKII-hMDR1-NKI cells these values are within twofold. We further visualized the microvilli in MDCKII-hMDR1-NKI and Caco-2 cells using three-dimensional structured illumination super-resolution microscopy and found that the microvilli in Caco-2 cells are taller and more densely packed than those in MDCK-hMDR1-NKI cells. We hypothesized over 10 years ago that only P-gp at the tips of the microvilli contribute significantly to efflux activity, whereas the remaining P-gp are involved in a futile cycle of efflux of amphipathic drugs from the microvillus membrane, followed by their reabsorption into the same or nearby microvillous membranes. The difference between the levels of total and efflux active P-gp in Caco-2 cells can be explained by the more densely packed microvilli in Caco-2 cells, which would lead to a substantial fraction of P-gp not contributing to final release of drug into the apical chamber. Our results suggest that the effect of microvilli morphology differences between in vitro and in vivo systems must be considered when scaling transporter activity for efflux transporters of amphipathic compounds, for example, P-gp.
Introduction
It is now well established that, in addition to drug-metabolizing enzymes, drug transporters also play a critical role in drug absorption, distribution, and elimination (Giacomini et al., 2010). Drug transporters affect drug concentrations in tissues and cells and therefore may be an important determinant of drug efficacy and safety. In addition, inhibition or induction of transporter activity can lead to drug-drug interactions.
Cytochrome P450-mediated hepatic drug metabolism in vivo can be predicted based on in vitro measurements of Km and Vmax, together with an intersystem extrapolation factor, which scales the in vitro Vmax to the appropriate in vivo Vmax. This intersystem extrapolation factor is used to take into account the differences in enzyme activity in vitro and in vivo (Proctor et al., 2004). At present, in vitro to in vivo extrapolation of transporter kinetic data often requires compound-dependent scaling factors (Harwood et al., 2013; Zamek-Gliszczynski et al., 2013). The reasons for this are currently not well understood and may be multiple.
Transporter kinetic parameters are typically generated using Michaelis-Menten approximations. Importantly, in vitro to in vivo extrapolation of kinetic parameters assumes that the in vitro Km is system independent and that the Vmax can be scaled based on total transporter expression levels, rather than transporter activity. Experimental evidence that these assumptions are mechanistically valid is typically lacking.
We have demonstrated previously for P-glycoprotein (P-gp) that Michaelis-Menten approximations are not always valid when applied to drug transport (Bentz et al., 2005). We developed a structural mass action kinetic model for P-gp–mediated transport, which is also a diagnostic tool for the detection of other transporters involved in transcellular transport (Acharya et al., 2008; Agnani et al., 2011; Lumen et al., 2013; Meng et al., 2016a).
Using this model, elementary rate constants (on-rate constant k1, off-rate constant kr, and efflux-rate constant k2) governing binding to and efflux from P-gp were obtained for several substrates and have been shown to be approximately the same in Caco-2 and MDCK-hMDR1-NKI cells. In addition, the structural model can estimate the efflux active P-gp level. Meng et al. (2016a) estimated the pmol efflux active P-gp/cell monolayer of a transwell in MDCKII-hMDR1-NKI cells and Caco-2 cell lines and showed that efflux active P-gp is about fivefold higher in MDCK-hMDR1-NKI cells compared with Caco-2 cells. With the recent development of protein quantification techniques, that is, targeted liquid chromatography–tandem mass spectrometry (LC-MS/MS), absolute levels of P-gp in the cell membrane can now be measured (Prasad and Unadkat, 2014). This allows us to explore whether P-gp total expression level is a good surrogate of its activity.
In the current work, we compared expression of total P-gp in Caco-2 and MDCK-hMDR1-NKI cell monolayers by Western blot and determined the absolute amount of P-gp in these monolayers by LC-MS/MS. We found that the total amount of P-gp in the cell monolayers was about the same, demonstrating a significant difference between total and efflux active P-gp. In fact, in Caco-2 cells, the pmol P-gp/cell monolayer of a transwell measured by LC-MS/MS was eight- to 12-fold higher than the fitted efflux active P-gp, whereas the difference between the measured and fitted values for MDCKII-hMDR1-NKI cells was less than twofold higher.
We speculated previously that only P-gp at the tips of microvilli contributes to efflux activity (Tran et al., 2005). An amphipathic compound effluxed at the bottom of a microvillus would randomly diffuse in the aqueous space between the microvilli and most likely be reabsorbed back into the microvillus membrane for another round of efflux. In this case, the morphology of microvilli could have an impact on the level of efflux active P-gp compared with total P-gp: the denser the microvilli, the smaller the fraction of efflux active P-gp. Simulations shown in Supplemental Figs. 3 and 4 have shown this.
This futile cycle could be seen wasteful from an evolutionary perspective, for example, why not tether the P-gp to the tips of the microvillus? Current work is showing that mouse enterocytes bleb vesicles from microvillus tips for many physiologic purposes (Shifrin et al., 2013). Thus, anchoring P-gp to the microvillus tip, then severing it from that anchor for the blebbing function and then reanchoring more P-gp to the tip is evidently more evolutionarily expensive than simply having all P-gp defusing over the microvillus, mostly efflux futile, but efflux active at the tips.
To test the question of whether microvilli morphology could explain the difference between total P-gp and efflux active P-gp, we visualized the microvilli in MDCKII-hMDR1-NKI and Caco-2 cells whose transport kinetics had been analyzed (Meng et al., 2016a), using three-dimensional structured illumination microscopy, a super-resolution microscopy method. We found that the microvilli in Caco-2 cells are indeed more densely packed than those in MDCK-hMDR1 cells. This finding most likely explains why the level of efflux active P-gp is lower in Caco-2 cells than in MDCK-hMDR1 cells, whereas their total P-gp was about the same.
Materials and Methods
Compounds and Reagents.
The Madin-Darby canine kidney II cell line overexpressing human MDR1 (MDCKII- hMDR1-NKI) was purchased from The Netherlands Cancer Institute (Amsterdam, The Netherlands). Caco-2 cells, which are human epithelial colorectal adenocarcinoma cells, were obtained from the American Type Culture Collection (Manassas, VA). Dulbecco’s modified Eagle’s medium with 25 mM HEPES, high glucose (4.5 g/L), L-glutamine, pyridoxine hydrochloride, without sodium pyruvate, and with phenol red, fetal bovine serum, minimal essential medium nonessential amino acids, and penicillin-streptomycin were from Thermo Fisher Scientific (Rockford, IL). Transwell 12-well plates with polycarbonate insert (0.4 µM pore size and 12 mm in diameter) were obtained from Corning Life Sciences (Acton, MA). Formic acid, acetonitrile (grade), bovine serum albumin, ammonium bicarbonate, iodoacetamide, and dithiothreitol were purchased from Sigma-Aldrich (Steinheim, Germany). The ProteoExtract Native Membrane protein extraction kit was purchased from Calbiochem (Temecular, CA). The synthetic P-gp peptides were purchased from Sigma-Aldrich, whereas their corresponding stable isotope-labeled (SIL) variants were purchased from Bachem (Torrance, CA). The protein quantification bicinchoninic acid kit was purchased from Pierce Biotechnology (Rockford, IL).
Cell Monolayer Culture and Membrane Protein Extraction.
MDCKII-hMDR1-NKI and Caco-2 cells were maintained in culture media (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 50 U/ml penicillin, 50 µg/ml streptomycin, and 0.1 mM minimal essential medium nonessential amino acids) and were kept at 37°C in 5% CO2. At 70–80% confluency, cells were washed twice with phosphate-buffered saline (PBS) and trypsinized with 0.25% trypsin/EDTA. MDCKII-hMDR1-NKI cells were split twice per week at a ratio of 1:10, whereas Caco-2 cells were split once per week at a ratio of 1:20. MDCKII-MDR1-NKI and Caco-2 cells were seeded on polycarbonate membrane inserts at a density of 2 × 105/cm2 and 2.5 × 105/cm2, respectively. The medium was changed every 2–3 days. MDCKII-hMDR1-NKI cells were grown for 4 days, whereas Caco-2 cells were maintained for 21 days before membrane protein extraction and immunohistochemistry.
Membrane protein was extracted using the Calbiochem native membrane protein extraction kit, according to the manufacturer’s suggested protocol with slight modifications. Briefly, cell monolayers were lysed in extraction buffer I of the extraction kit containing the appropriate amount of protease inhibitor cocktail. Cell monolayers were incubated for 10 minutes at +4°C under gentle agitation. The supernatant containing the cytosolic protein was removed, and the respective pellets were resuspended in extraction buffer II of the extraction kit with protease inhibitor cocktail. After 30-minute incubation at +4°C with gentle agitation, the supernatant containing the membrane protein was collected and stored at −80°C for future analysis. Exactly the same extraction procedure using buffer II was repeated four times on the same cell monolayer (Supplemental Fig. 1). Four rounds of extraction were required to make sure that essentially all of the membrane protein has been extracted. The same volume of extraction buffer I and II per transwell was used in MDCKII-hMDR1-NKI and Caco-2 cell lines. The sample from each round of extraction was combined, and the total protein concentration was determined using a bicinchoninic acid protein assay kit.
Western Blot.
The combined membrane protein extract from the previous step was directly used in the Western blot without any modification of protein concentration. The same volume of membrane protein extract from MDCKII-hMDR1-NKI and Caco-2 cell lines containing 10–30 μg protein was separated on precast 7.5% Bis-Tris gels (Bio-Rad, Hercules, CA) and transferred to a polyvinylidene fluoride membrane (Bio-Rad) at 200 mA for 2 hours. After being blocked with 5% nonfat dry milk powder in Tris-buffered saline/0.1% Tween 20 buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) for 30 minutes, membranes were incubated with primary antibody C219 (Signet Laboratories, Dedham, MA) against P-gp, diluted 1:200, at 4°C overnight, followed by washing in Tris-buffered saline/0.1% Tween 20. Membranes were further incubated with secondary antibody anti-mouse IgG horseradish peroxidase (Dako, Glostrup, Denmark), diluted in 1:1000, at room temperature for 2 hours. Protein bands were visualized by chemiluminescence (ECL plus Western Blotting Substrate System; Thermo Fisher Scientific), according to the manufacturer’s instruction, and were developed using HyBlot CL film (Denville, Holliston, MA). The band intensity was quantified by software ImageJ (W. Rasband, National Institutes of Health).
Peptide Selection, Sample Preparation, and Trypsin Digestion of Membrane Proteins.
Two unique tryptic-derived signature peptides were selected for quantification of P-gp based on previously reported criteria (Kamiie et al., 2008; Zolk and Fromm, 2011) and literature reports (Miliotis et al., 2011; Zhang et al., 2011). The selected signature peptides for P-gp were AGAVAEEVLAAIR (amino acid residues from 250 to 262) and NTTGALTTR (amino acid residues from 809 to 817), which are located at the fourth and eighth cytoplasmic loops, respectively.
An aliquot of 100 µL extracted membrane protein from the previously discussed extraction procedure from each cell line was diluted 1:1 with 100 mM ammonium bicarbonate solution in a 1.5 mL Eppendorf tube. The membrane samples then were reduced using 10 mM dithiothreitol at 50°C for 30 minutes and alkylated with 15 mM iodoacetamide in the dark at room temperature for 45 minutes to prevent the free sulfhydryls from reforming disulfide bonds. The samples then were delipidated by adding cold methanol (600 µL), chloroform (150 µL), and water (450 µL) and vortexed for 1 minute sequentially. Subsequent centrifugation at 15,000 rpm for 5 minutes at 4°C resulted in a separation into a top aqueous and bottom organic layer. A very thin protein film was observed in the interface of the two phases. The upper aqueous layer was removed carefully without disturbing the protein film. A total of 450 µl cold methanol was added further into the sample and then followed by centrifugation at 15,000 rpm for 5 minutes at 4°C. The supernatant was discarded carefully. The pellets were briefly dried in a vacuum centrifuge. The pellets were resuspended in 60 µL digestion buffer (100 mM ammonium bicarbonate and acetonitrile in a ratio of 9:1) by intermittent sonication on ice. After addition of internal standard, 0.5 pmol/µL SIL P-gp proteotypic peptides (AGAVAEEV[15N113C6]LAAIR and NTTGAL[15N113C6]TTR), the samples were digested with trypsin (Promega, Madison, WI; 1:20, trypsin:protein) at 37°C overnight in a final volume 70 µL. The mass ratio of trypsin to protein was 1:20. The digestion was discontinued by addition of formic acid (3 µL, 50% v/v), and the sample was centrifuged at 15,000 rpm for 5 minutes at 4°C. Finally, the clear supernatant was evaporated to dryness using a vacuum centrifuge for 3 hours at 50°C. The samples were reconstituted in 60 µL 0.1% (v/v) formic acid prior to LC-MS/MS analysis. All samples were digested and analyzed using LC-MS/MS in duplicate. All sample preparation and digestion steps were formed using Protein Lobind tubes (Eppendorf, Hamburg, Germany).
LC-MS/MS Analytical Method Parameters.
LC-MS/MS analyses were conducted on an API4000 triple-quadrupole mass spectrometer coupled with Ultra-Performance Liquid Chromatography system (Sciex, Concord, Ontario, Canada). The liquid chromatography column used for peptide separation and elution was a 2.1 × 50-mm C18 column (Acquity UPLC BEH C18, 1.7 µm size beads, 130 Å pore size; Waters, Milford, MA). Mobile phase A was water with 0.1% v/v formic acid, and mobile phase B was 50% acetonitrile/50% isopropanol. The chromatographic separation was achieved by a linear gradient starting from 5% B and progressing to 85% B over a period of 7 minutes. A sample volume of 10 µL was injected onto the LC column at a flow rate of 0.5 mL/min. The precursor-to-product transition for the quantification of the peptide NTTGALTTR represented the doubly charged precursor ion [M + 2H]2+ (m/z 467.8) and the singly charged product y7 ion with m/z 719.4. Regarding peptide AGAVAEEVLAAIR, the doubly charged precursor ion [M + 2H]2+ (m/z 635.4) gives rise to the singly charged product ions y9 with m/z 971.5. The instrument settings of the API4000 triple-quadrupole mass spectrometer were as follows: ion spray voltage, 4 kV; temperature, 400°C; declustering potential, 50 V; collision energy, 38 V; entrance potential, 10 V; and collision cell exit potential, 11 V.
The calibration curve standards were prepared by spiking both the NTTGALTTR and AGAVAEEVLAAIR over the range of 780 pM to 50,000 pM and the SIL peptides (internal standard) at a fixed concentration of 50 nM into a surrogate matrix (1 µg/µL tryptically digested bovine serum albumin). Assay accuracy and precision were determined in triplicate at two different quality control concentrations of 1000 and 10,000 pM peptide in surrogate matrix and then subjected to tryptic digestion. Data were processed by integration of the appropriate peak areas generated from the multiple-reaction monitoring (MRM) chromatograms of either the AGAVAEEVLAAIR and NTTGALTTR peptides or the corresponding stable label peptides using Analyst 1.4.2 (Applied Biosystems, Foster City, CA). The peak area ratio of the peptides to the corresponding SIL peptide (y) was plotted against the nominal concentration (Supplemental Fig. 2).
LC-MS/MS Assay.
The calibration curves were linear across the concentration range of 780–50,000 pM for both P-gp peptides with correlation coefficients (r2) of greater than 0.99 (Fig. 2). The retention time for P-gp peptide standard and SIL internal standard was 2.5 minutes for AGAVAEEVLAAIR and 1.5 minutes for NTTGALTTR, respectively. Accuracy and precision in the quantification of the quality control samples were acceptable (CV < 15%) at two different concentrations. Trypsin digestion time was chosen by determining the incubation time at which there was no further increment in the yield of peptide (data not shown). Because purified P-gp protein standard is not available, the analytical method was based on the assumption that P-gp is completely extracted from the cell lysates and completely digested its corresponding peptides.
Immunohistochemistry.
Cell monolayers were rinsed twice with PBS before fixing with 3% w/v paraformaldehyde for 15 minutes. Fixed monolayers were permeabilized with 0.1% v/v Triton X-100 in PBS for 15 minutes and rinsed with PBS. Samples were blocked for 30 minutes with 2% w/v bovine serum albumin in PBS to prevent nonspecific binding. Then samples were incubated with primary mouse anti-human MDR1 antibodies: 5 µg/ml MRK16 (Enzo Life Science, Farmingdale, NY) in blocking solution for 2 hours at room temperature. Cells were rinsed and washed three times with blocking solution to remove unbound primary antibody. Cells were further incubated with a solution of the secondary Alexa Fluor 594–labeled donkey anti-mouse IgG (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA) in blocking buffer for 1 hour, followed by Alexa Fluor 488 phalloidin (Thermo Fisher Scientific, Rockford, IL) staining of filamentous actin for 30 minutes. Inserts were washed with PBS before the filter was excised and mounted on a slide using VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA)
Three-Dimensional Structured Illumination Microscopy.
Super-resolution imaging was performed using the DeltaVision OMX V4 microscope (GE Healthcare) with a PlanApoN 60×/1.42 N objective (Olympus) and immersion oil with a refractive index of 1.516. Alexa Fluor 488 was excited by a 488 nm solid-state laser, and its fluorescence emission was collected between 504 and 552 nm. Alexa Fluor 594 was excited by a 568 nm solid-state laser, and its fluorescence emission was collected between 590 and 628 nm. A z-step size of 0.125 µm was used, and images were sequentially acquired with sCMOS PCO.edge cameras (PCO-Tech). Raw images containing three-dimensional structured illumination microscopy illumination patterns were reconstructed, and the channels were aligned with softWoRx software (version 6.5.1; GE Healthcare). Channel alignments in x, y, and rotationally around the z-axis were obtained using predetermined shifts measured from a registration calibration slide. Channels were aligned in z using 100-nm Tetraspeck fluorescent microspheres (Thermo Fisher Scientific). Imaging settings were identical between MDCKII-hMDR1-NKI and Caco-2 cells.
Image Analysis.
Maximum intensity projections of image stacks and orthogonal views were generated with FIJI/ImageJ open source imaging software (http://fiji.sc).
Results
Expression of P-gp by Western Blot.
The expression of P-gp in MDCKII-hMDR1-NKI and Caco-2 cell monolayers was determined by SDS-PAGE and Western blot analysis of the membrane protein extract. Figure 1A shows the P-gp protein detected after SDS-PAGE of identical volumes of membrane protein extract. Figure 1B shows the average densitometric value of the P-gp bands from three independent rounds when the sample loading volumes are 40 μL. The results indicate that MDCKII-hMDR1-NKI and Caco-2 cells contain approximately the same amount of P-gp in 40 μL membrane protein extract. Because the same volume of buffer was used in the two cell lines during monolayer extraction, the results indicate that P-gp expression per cell monolayer of a transwell was about the same in the two cell lines.

Comparison of P-gp expression in confluent MDCKII-hMDR1-NKI and Caco-2 cell monolayer. In (A), lanes 1 and 3, MDCKII-NKI-hMDR1; lanes 2 and 4, Caco-2. Lanes 1 and 2 contain 40 µL membrane protein extract, whereas lanes 3 and 4 contain 20 µL membrane protein extract. In (B), densitometric values of Western blot bands from 40 µL membrane protein extract were quantified by ImageJ, and the means ± S.E.M. were from three independent experiments. Expression of P-gp protein of 40 µL membrane protein extract was almost the same between MDCKII-hMDR1-NKI and Caco-2 cells without significant difference (Student pair t test, P value = 0.09)
LC-MS/MS Quantification of P-gp in MDCKII-hMDR1-NKI and Caco-2 Cell Monolayers.
The LC-MS/MS method described above was applied for the determination of P-gp protein levels in the membrane fractions extracted from MDCKII-hMDR1-NKI and Caco-2 cell monolayers, cultured for 4 days and 21 days, respectively. The specific MRM transitions based on the chromatographic retention time and mass spectrometry spectrum were used to detect and quantify the P-gp proteotypic peptide by LC-MS/MS in MRM mode. As shown in Table 1, the membrane P-gp was determined in 2 U that corresponded to the following: 1) pmol P-gp per cell monolayer of a 12-well transwell plate and 2) pmol P-gp per mg membrane protein. The first unit is needed for comparison with our fitted efflux active P-gp values, whereas the second unit is the one commonly used in the literature. The Caco-2 cells contained approximately twice as much cell protein per transwell as the MDCKII-hMDR1-NKI cells. The quantification results from the two P-gp signature peptides were close with minor difference (1.5-fold). Assuming that the difference was due to different degrees of trypsin digestion, the results used for comparison between the two cell lines in Table 2 were based on peptide 1. Consistent with the results from the Western blot, the expression level of P-gp per cell monolayer of a transwell was very similar between the two cell lines, with a ratio of MDCK/Caco-2 of approximately 0.8 (Table 2).
P-gp content as determined by LC-MS/MS in MDCKII-hMDR1-NKI and Caco-2 cell monolayers
Comparison of efflux active P-gp with total P-gp in MDCKII-hMDR1-NKI and Caco-2 cells
Comparison of Efflux Active P-gp with Total P-gp in MDCKII-hMDR1-NKI and Caco-2 Cells.
Table 2 shows that the ratio of total P-gp per cell monolayer of a transwell between MDCKII-hMDR1-NKI and Caco-2 cells was about 0.8 determined by LC-MS/MS. In addition, the fitted efflux active P-gp (pmol/cell monolayer of a transwell), obtained from (Lumen et al., 2013) and (Meng et al., 2016a), is also shown in Table 2. Fitted efflux active P-gp was about fivefold higher in MDCK-hMDR1 cells than in Caco-2 cells. Table 2 also shows that total P-gp in Caco-2 cells is eight- to 12-fold greater than efflux active P-gp, whereas the values in MDCKII-hMDR1-NKI cells are similar.
Characterization of Microvillus Morphology in MDCKII-hMDR1-NKI and Caco-2 Cells.
Because our kinetic analysis only measures those P-gp that efflux drug into apical chamber, which are defined as efflux active P-gp, we had hypothesized that only the P-gp expressed at the tips of microvilli are efflux active (Tran et al., 2005). If this is the case, morphology and density of microvilli may have a significant impact on the fraction of P-gp that is efflux active. Therefore, we used super-resolution microscopy to investigate the morphology of microvilli on MDCKII-hMDR1-NKI and Caco-2 cells, grown in transwells under the exact same conditions as used for the P-gp transport experiments (Fig. 2). Microvilli were visualized by staining filamentous actin with phalloidin, whereas P-gp was visualized by indirect immunofluorescence. The microvilli of Caco-2 cells were more densely packed and more regularly shaped than on MDCKII-hMDR1-NKI cells (Fig. 2, A and B). In addition, although all of the Caco-2 microvilli were perpendicular to the cell surface, some of the microvilli of MDCKII-hMDR1-NKI cells were not. P-gp was not found in the cytoplasm, but localized almost exclusively in the apical microvilli-containing region of the cells (data not shown). Notably, in both cell lines, there were cell-to-cell variations in the levels of P-gp expression, as previously observed in Caco-2 cells (Fig. 3) (Hunter et al., 1993; Anderle et al., 1998).
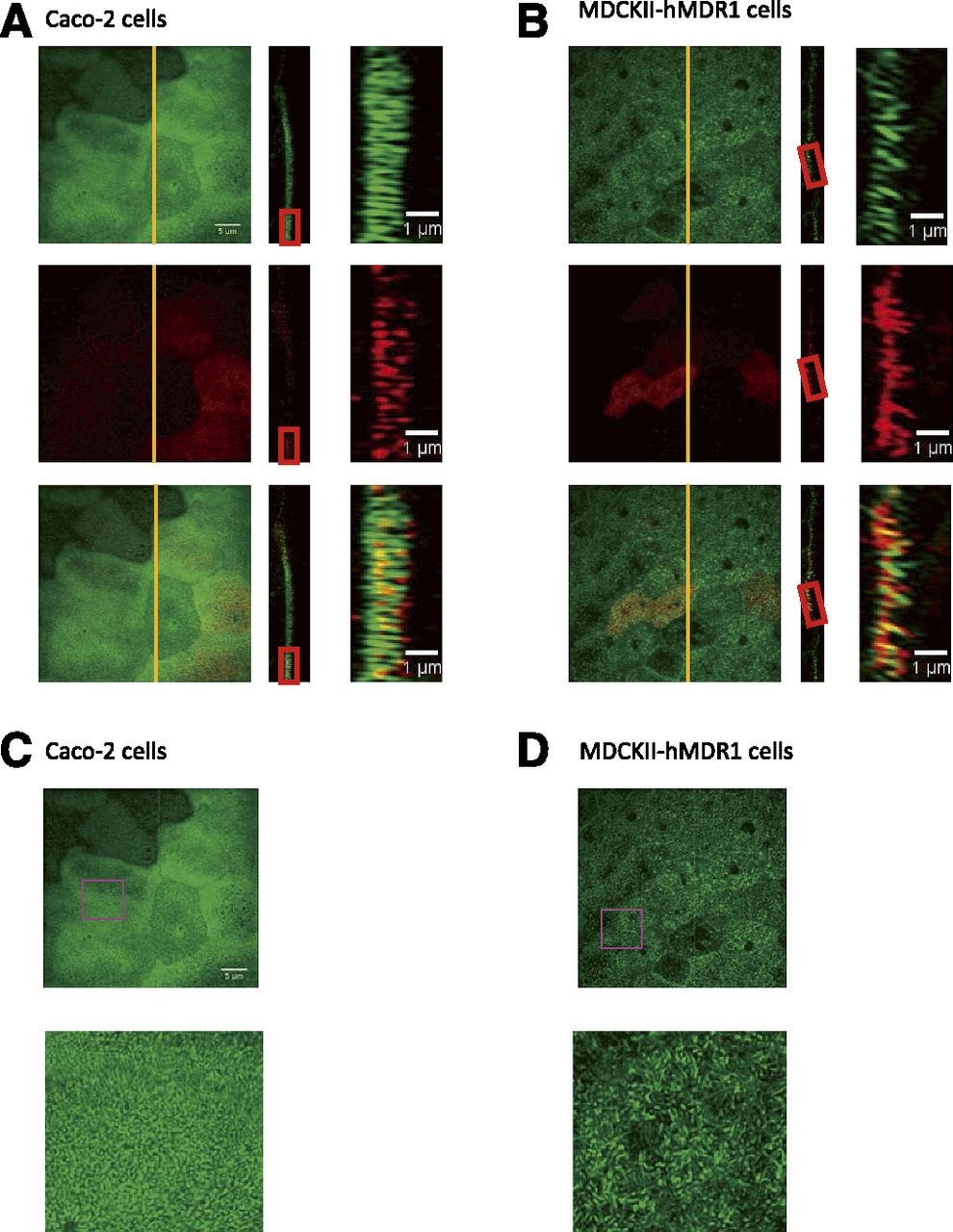
Characterization of microvilli morphology and P-gp localization in confluent MDCKII-hMDR1-NKI and Caco-2 cell monolayers by three-dimensional structured illumination microscopy. Representative images of microvilli, as seen by filamentous actin staining (green) and P-gp staining (red) in Caco-2 cells (A, C) and MDCKII-hMDR1-NKI cells (B, D). (A and B) Left panels show the maximum intensity z-projections; rectangle panels next to the left panels show the orthogonal (yz) views obtained from the vertical yellow lines shown in the z-projections. Panels on the right are high magnification images obtained from the red rectangles. (C and D) All panels show the maximum intensity z-projection; the panels of second row show the high magnification images obtained from the purple square.
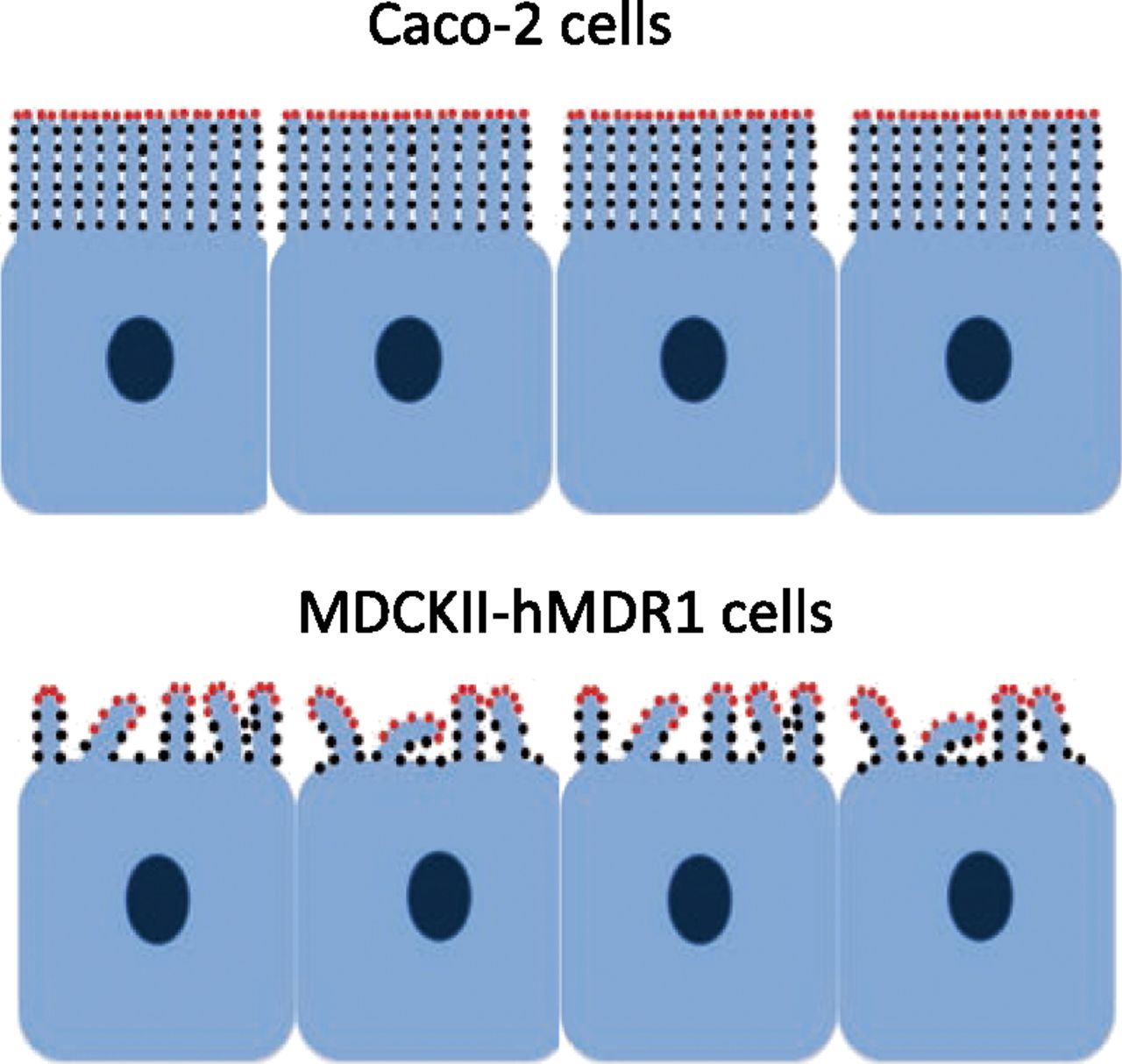
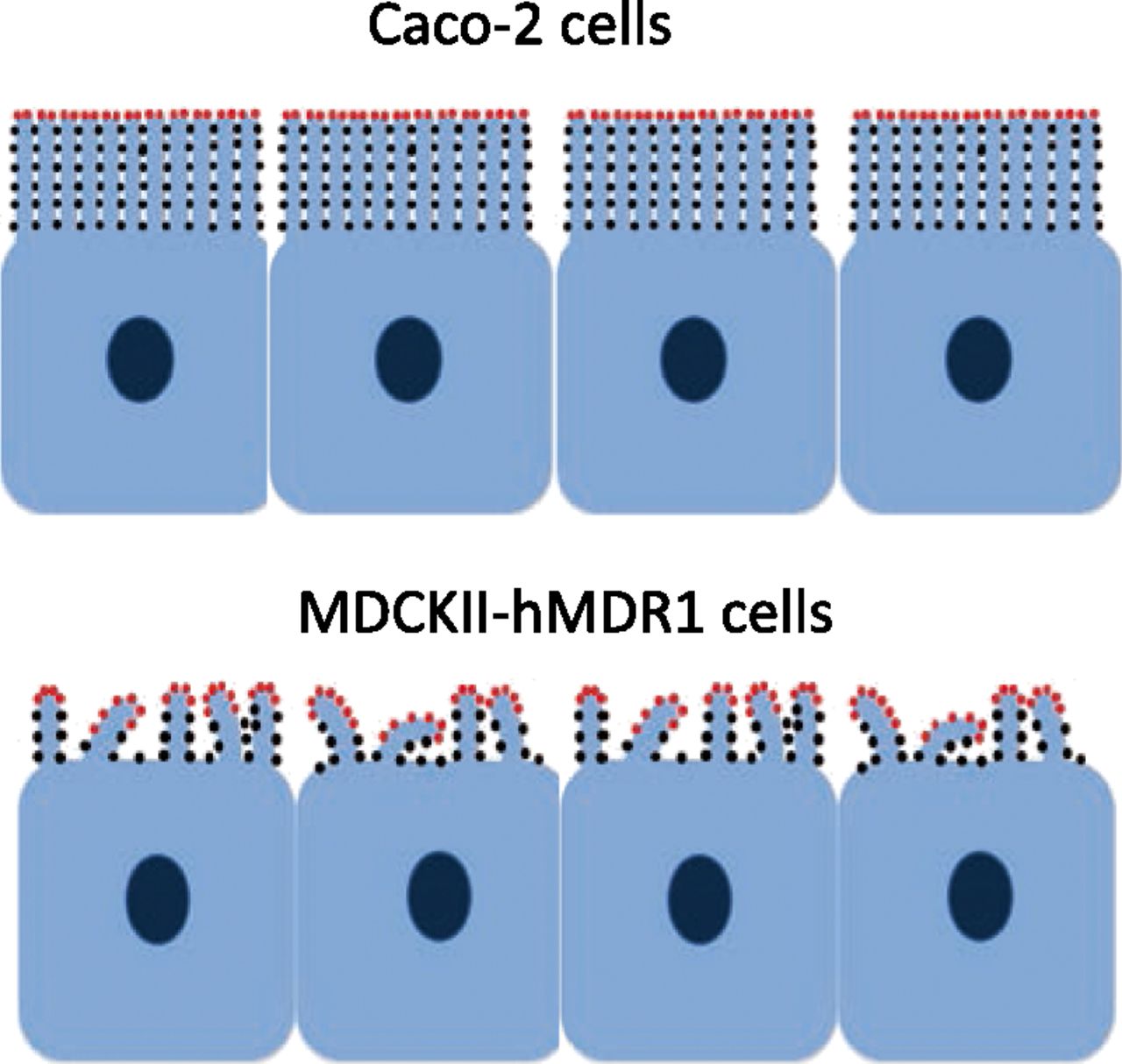
Cartoon figures of Caco-2 and MDCKII-hMDR1-NKI cells with microvilli and P-gp (dots). Red dots represent efflux active P-gp that can send their drug directly to apical chamber, whereas black dots represent efflux inactive P-gp. The morphology of microvilli was drawn approximately based on results from super-resolution microscopy, as shown in Fig. 2. In mouse small intestine, there is about 50 nm between adjacent microvilli, with some variability in the size of the aqueous space between them (Crawley et al., 2014). Microvilli of Caco-2 cells are more packed and regular than the ones in MDCKII-hMDR1-NKI cells. Differently from Caco-2 cells, microvilli on MDCKII-hMDR1-NKI cells are not all standing straight up; many of them are bent. Previously we proposed that the amphipathic P-gp substrate drug released at the base of a microvillus would be interacting with the same or a neighboring microvillus and the P-gp they contain (Bentz et al., 2005; Tran et al., 2005). Only the P-gp at the tips and at other apical chamber-exposed surfaces of the MDCKII-hMDR1-NKI bent microvilli can efflux the drug directly into the apical chamber with higher probability. According to this hypothesis, a larger fraction of total P-gp would be efflux active in MDCKII-hMDR1-NKI (red dots) when the total P-gp levels are similar among the two cell lines.
Discussion
The lack of transporter-specific probe substrates and inhibitors hampers clinical investigation of the contributions of individual transporters to drug absorption, distribution, metabolism, and excretion and mechanistic extrapolation to the absorption, distribution, metabolism, and excretion of other transporter substrates or inhibitor drugs. Physiologically based pharmacokinetic modeling is increasingly used as a tool to probe the role of transporters in drug disposition. Michaelis-Menten kinetic parameters for drug-transporter interaction (Km and Vmax) are determined in vitro, either using primary cells (e.g., hepatocytes) or single transporter-overexpressing cells, for example, Caco-2 (Zamek-Gliszczynski et al., 2013). The rate of transporter-mediated clearance (Vmax/Km) in vivo is then predicted by scaling the Vmax based on relative transporter expression levels in vitro and in vivo. This assumes Km is the same in vitro versus in vivo and that the total transporter levels in vitro and in vivo are efflux active.
We have discussed previously the potential pitfalls in assuming that the Michaelis-Menten Km is system independent (Bentz et al., 2005; Bentz and Ellens, 2014; Meng et al., 2016a). In this study, we examine the assumption that total transporter equals efflux active transporter. Recently, it was reported that transport activity of breast cancer–resistant protein expressed in MDCK cells was highly correlated with protein expression level (Kumar et al., 2015), but that observation is not inconsistent with our results. A higher P-gp surface density at the microvilli tips would yield the same result.
We have previously fitted efflux active P-gp in MDCKII-hMDR1-NKI and Caco-2 cells using a structural mass action kinetic model (Tran et al., 2005; Agnani et al., 2011; Lumen et al., 2013; Bentz and Ellens, 2014). Meng et al. (2016a) demonstrated that the efflux active P-gp per cell monolayer of a transwell was about fivefold greater in MDCKII-hMDR1-NKI than in Caco-2 cells. To investigate the relationship between total P-gp and efflux active P-gp, we first compared total P-gp level in these two cell lines by Western blot, and our results implied that the total P-gp per cell monolayer of a transwell is nearly the same between MDCKII-hMDR1-NKI and Caco-2 cell lines.
Because Western blots were only semiquantitative due to the unavailability of pure P-gp protein, total P-gp level was further quantified using LC-MS/MS–based targeted proteomics, which has shown to be a promising approach for the determination of absolute level of transporter protein (Prasad and Unadkat, 2014). In this study, two unique P-gp signature peptides were selected for the quantification. From the results shown in Table 1, we observed a minor (up to 1.5-fold) but systematic difference (p = 0.023 in both MDCK and Caco-2) in P-gp quantification between two different signature peptides. Because peptide AGAVAEEVLAAIR yielded the higher amount of P-gp in both cell lines, we assume that this difference was due to different trypsin digestion efficiencies.
Therefore, the results that are used for comparison of absolute amounts of P-gp in the two cell lines are based on peptide AGAVAEEVLAAIR. In agreement with the Western blot results, the pmol P-gp per cell monolayer of a transwell is very similar between the two cell lines. We also calculated pmol P-gp per mg membrane protein, which is a unit commonly used in the literature. Our results are comparable to reported literature values for Caco-2 cells (Miliotis et al., 2011) and MDCKII-MDR1 cells (Zhang et al., 2011).
Other data using Western blots show differences between Caco-2 and MDCK-hMDR1 cell lines, for example (Taub et al., 2005). We ascribe this to the finding in Bentz et al. (2013) that the IC50 values among just the Caco-2 cells varied about 150-fold for all inhibitors used, and Lumen et al. (2013) showed that IC50 values were proportional to efflux active P-gp. Ultimately, there is no reason to assume that the Caco-2 cells in any two laboratories will express the same amount of efflux active P-gp because it depends upon culture conditions (Hayeshi et al., 2008) and upon total expressed P-gp and microvilli morphology.
The total pmol P-gp per cell monolayer of a transwell measured by LC-MS/MS is 8–12 times greater than the fitted efflux active pmol P-gp per cell monolayer of a transwell for Caco-2 cells, whereas the values are similar for MDCKII-hMDR1-NKI cells. This result implies that, at least for Caco-2 cells, a significant minority of the P-gp is efflux active.
The question of whether measuring P-gp mRNA levels could estimate relative protein expression levels, as opposed to using LC-MS/MS, was addressed. Table 3 shows the comparison of the ratios of P-gp mRNA to P-gp measured by LC-MS/MS per well. There is ninefold more P-gp mRNA per pmol P-gp in MDCKII-hMDR1-NKI cells than in Caco-2 cells. Without a greater number of data points from many more MDCKII-hMDR1 and Caco-2 cells, there seems little reason to assume any simple relationship.
Comparison of efflux active P-gp with total P-gp mRNA in MDCKII-hMDR1-NKI and Caco-2 cells
Kits used for isolation of membrane proteins, such as used in the current work, do not distinguish between plasma membrane and intracellular membranes. Although several commercial kits claimed to isolate plasma membrane protein, it has been shown recently that these kits either yielded very little plasma membrane or failed to separate plasma membrane from other intracellular membranes (Kumar et al., 2015). In our calculations we assume that all P-gp extracted from the MDCKII-hMDR1-NKI and Caco-2 cells was present in the apical membrane. Although P-gp has been found in intracellular membranes in certain cell lines (Arancia et al., 2001), it has shown to be almost exclusively expressed in the apical membrane of MDCK and Caco-2 cells (Anderle et al., 1998; Hutter et al., 2014). Evidence for this was also obtained in our Caco-2 and MDCKII-hMDR1-NKI micrographs, where P-gp staining was essentially limited to the apical membrane (data not shown). Therefore, the difference between total and efflux active P-gp in Caco-2 cells cannot be explained by a speculation that a large proportion of P-gp is localized intracellularly. Even if this were the case, the efflux active P-gp is responsible for tissue clearance by P-gp.
We hypothesized that only the P-gp at the tips of microvilli is efflux active (Tran et al., 2005). The prediction that efflux active P-gp depends upon microvilli morphology was supported by simulations (Supplemental Figs. 3 and 4). If true, the fraction of efflux active P-gp would be affected by the morphology of microvilli: the more densely packed the microvilli, the smaller the fraction of efflux active P-gp. Using super-resolution microscopy to visualize microvilli (via staining for actin), we demonstrated that microvilli of Caco-2 cells are very densely packed, whereas in MDCKII-hMDR1-NKI cells they are located much farther apart. In addition, the microvilli found in MDCK cells, which are not all perpendicular to the cell surface, allow more drugs to be effluxed into apical chamber by P-gp because a larger fraction of the microvilli surface area is exposed to the apical chamber.
This is consistent with published electron micrographs of Caco-2 and MDCK cells (Butor and Davoust, 1992; Gill et al., 2008; Nicolaou et al., 2012). Using super-resolution microscopy, it is possible to obtain data on multiple proteins in the same image. In addition to visualization of actin in microvilli, we also visualized P-gp through staining with fluorescent antibody. P-gp is distributed in the apical membrane and spread evenly across the microvilli membranes. The microscopy data are consistent with the hypothesis that the difference between total and efflux active P-gp in Caco-2 cells is due to microvilli morphology. The super-resolution microscopy has allowed us to measure microvilli morphology for the same cells grown identically for transport kinetics. This eliminates much of the ambiguity in the results found for laboratory-to-laboratory variations in IC50 values found in Bentz et al. (2013).
P-gp is an ATP-binding cassette transporter and uses the energy from ATP binding and hydrolysis to efflux its substrate. One may speculate that the difference between efflux active P-gp and total P-gp can be due to different ATP concentration and regulation in the two cell lines. However, this cannot be true because, in our companion paper (Meng et al., 2016a), we showed that the efflux rate constants of P-gp, which reflect the activity of ATP, were very similar between MDCKII-hMDR1-NKI and Caco-2 cells, and constant over 6 hours. It is also unlikely that the difference in efflux active P-gp between the two cell lines is due to a different localization pattern of P-gp on microvilli. According to Fig. 2, A and B, expression of P-gp along with actin is very similar in both cell lines.
As mentioned above, protein expression is assumed to be the closest surrogate of transporter activity and is commonly used as a scaling factor for Vmax in PBPK modeling. However, we have demonstrated that the level of efflux active P-gp can be affected by the morphology of microvilli. That is to say, the extrapolation of Vmax with this scaling method should only be used between two cell lines with similar microvilli structure, a difficult thing to quantitatively demonstrate. For example, the microvilli morphology of MDCKII-hMDR1-NKI cells that we observed in this work is approximately similar to that in bile canaliculi from mouse liver (Crawford et al., 1997) and quite different from the one in intestinal epithelium of guinea pigs (Takeuchi and Sprinz, 1967). The scaling of Vmax using total expression levels would be more accurate when the extrapolation is from MDCKII-hMDR1-NKI cells to bile canaliculi than to small intestinal cells in vivo.
A fair question is whether the microvilli morphologies shown in this work reflect those of healthy human tissue. There will certainly be differences. However, the point is that in vivo absorption depends upon efflux active P-gp, which depends upon microvilli morphology, as well as the contribution of a basolateral uptake transporter. Our kinetic model can measure those contributions to the absorption, as described in Meng et al. (2016a).
Conclusions
We found that in Caco-2 cells total P-gp is much greater than efflux active P-gp, whereas in MDCKII-hMDR1-NKI cells the values are approximately similar. The most likely reason for this is a significant difference in microvilli morphology between the two cell lines. Although P-gp is evenly distributed along the entire microvillous membrane in both cell types, the microvilli in Caco-2 cells are more densely packed than those in MDCKII-hMDR1-NKI cells, leading to a substantial fraction of P-gp in Caco-2 cells not contributing to final release of drug into the apical chamber, but instead being involved in a futile cycle of drug efflux, followed by rapid reabsorption of the amphipathic P-gp substrates. The effect of microvilli morphology differences between in vitro and in vivo systems must be considered when scaling transporter activity for P-gp and possibly other efflux transporters.
Acknowledgments
The authors thank Dr. Elias Spiliotis, Dr. Nianli Sang, Dr. Chengqin Yin, and Dr. Xiaobo Bai (Biology Department, Drexel University) for expert assistance with the experiments and Dr. Michael O’Connor (Biodiversity, Ecology, and Earth Sciences, Drexel University) for their statistical analysis. Super-resolution microscopy was performed in the Cell Imaging Center, Drexel University. The authors also thank Neha Manjari Akella, a student at Drexel University College of Medicine who contributed during the early stage of LC-MS/MS protocol design.
Authorship Contributions
Participated in research design: Meng, Le Marchand, Agnani, Szapacs, Ellens, Bentz.
Conducted experiments: Meng, Le Marchand, Agnani.
Performed data analysis: Meng, Le Marchand, Agnani, Szapacs, Ellens, Bentz.
Wrote or contributed to the writing of the manuscript: Meng, Le Marchand, Agnani, Szapacs, Ellens, Bentz.
Footnotes
- Received June 21, 2016.
- Accepted November 7, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MRM
- multiple-reaction monitoring
- PBS
- phosphate-buffered saline
- P-gp
- P-glycoprotein
- SIL
- stable isotope-labeled
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}