


Abstract
Preincubation of a drug transporter with its inhibitor in a cell-based assay may result in the apparent enhancement of the inhibitory potency. Currently, limited data are available on potentiation of transporter inhibition by preincubation (PTIP) for clinically relevant solute-carrier transporters other than OATP1B1 and OATP1B3. Therefore, PTIP was examined systematically using OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2-K cell lines. IC50 values of 30 inhibitors were determined with or without 3 hours of preincubation, and compounds with a PTIP ≥2.5× were further characterized by assessing the time course of transport inhibition potency and cellular concentration. For each compound, correlations were calculated between highest observed PTIP and physicochemical properties. PTIP was prevalent among organic cation transporters (OCTs) and organic anion–transporting polypeptides (OATPs) but not among organic anion transporters (OATs) or multidrug and toxin extrusion transporters (MATEs), and most instances of PTIP persisted after controlling for toxicity and nonspecific binding. Occasionally, preincubation in excess of 2 hours was required to attain full inhibitory potency. For four drugs examined, preincubation had the potential to change the in vitro drug-drug interaction risk prediction from “no risk” to “risk” on the basis of current regulatory criteria. Molecular weight and LogD7.4, as well as the ratio of passive cellular accumulation and cellular uptake rate correlated with PTIP; thus, low cellular permeation and a slow build-up of unbound intracellular inhibitor concentration may contribute to PTIP. Taken together, our data suggest that PTIP is partly determined by the physicochemical properties of the perpetrator drug, and preincubation may affect the in vitro predicted drug-drug interaction risk for OCTs as well as OATPs.
Significance Statement During the development of a novel pharmaceutical drug, in vitro studies are conducted to assess the risk of potential adverse interactions between existing medications a patient may already be taking and the novel compound. The exact way these in vitro assays are performed may influence the outcome of risk assessment. Here we suggest that the interaction risk may be underestimated unless specific assay protocols are modified to include an additional incubation step that allows the test drug to accumulate inside the cells, and demonstrate that adding this step is particularly important for large and hydrophobic drug molecules.
Introduction
Uptake transporters involved in drug clearance accept a broad spectrum of substrates, and their contribution to cellular uptake is especially important for low-permeability compounds. Whenever transporter-mediated uptake becomes the rate-limiting step in drug clearance, inhibition of transport may lead to clinically relevant exposure increases (Lee et al., 2017). Therefore, health authorities recommend the routine investigation of new drug candidates as transport inhibitors at various stages of clinical development (EMA, 2012; FDA, 2017; PMDA, 2018). A classic example of transporter-mediated drug-drug interaction (DDI) is the interference of cyclosporin A (CsA) with the OATP-mediated uptake of statins into hepatocytes (Hirano et al., 2006). By inhibiting the active cellular uptake of statins, CsA restrains their access to their hepatic target and increases exposure of peripheral tissues; thus, it simultaneously limits the efficacy of statins and increases the potential for adverse effects (Neuvonen et al., 2006).
In vitro uptake inhibition assays are highly instrumental in predicting transporter-based DDIs; however, to yield physiologically relevant predictions, these models should mimic real-life drug exposure patterns. A putative perpetrator may be administered in the clinic for prolonged periods and attain steady-state concentrations in all accessible tissues before a victim drug is taken. In contrast, traditional in vitro assays comprise only a simultaneous incubation of the transporter with a probe substrate and the test drug. Coincubation allows only for immediate and direct interaction of the test drug with the transporter binding site and may hence underestimate DDI risk. Early evidence for this came from studies in which the inhibitory effect of a single dose of CsA on the hepatic clearance of bromsulphthalein persisted for at least 3 days in rats (Shitara et al., 2009), and preincubation before coincubation of OATP1B1 with CsA, compared with coincubation only, resulted in up to a 20-fold reduction in the measured IC50 values (Amundsen et al., 2010; Shitara et al., 2012). Recent literature indicates that additional compounds, including ritonavir, saquinavir, and anthraquinones, can exert a more potent inhibition of uptake transport activity upon preincubation (Amundsen et al., 2010; Shitara et al., 2013; Ma et al., 2015; Shitara and Sugiyama, 2017).
Although this potentiation of transport inhibition by preincubation (PTIP) may at first seem to be analogous to the time-dependent inhibition of drug-metabolizing enzymes, its mechanism would probably be different. It is hypothesized that such inhibitors can interact with the transporter on both the extracellular side (cis) and the cytosolic side (trans). PTIP is thought to arise when trans-inhibition is more potent than cis-inhibition, and intracellular accumulation of the inhibitor is slow (Shitara and Sugiyama, 2017). The time requirement of maximum inhibition is compound-specific and probably correlates with the membrane permeability of the inhibitor. Another potential source of apparent PTIP in in vitro assays may be nonspecific binding (NSB) of the inhibitor to plasticware and/or cells during preincubation. Lipophilic compounds like saquinavir, ritonavir, or CsA may saturate available nonspecific binding sites during the preincubation period, and no additional amounts of these inhibitors would be irreversibly lost during the transport step of the uptake assay.
Currently, inhibition assays for clinically relevant drug transporters other than OATPs are conducted without a preincubation step. The omission of this step can be considered a shortfall when attempting to mimic in vivo conditions in which an inhibitor is allowed to attain steady-state tissue concentrations prior to the administration of the test substrate. Most studies concerning PTIP have focused on OATPs, prompting regulatory agencies to recommend the inclusion of a preincubation step when OATP transport inhibition is being examined. Although sporadic studies have addressed the effect of preincubation on OATs (Ma et al., 2015) and OCTs (Arakawa et al., 2017), the lack of a systematic examination of non-OATP transporters has limited our knowledge of whether such mechanisms may be of additional clinical relevance.
In this study, the preincubation effect of 30 compounds was investigated on OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, as well as multidrug and toxin extrusion (MATE) 1 and MATE2-K operated in inverse (uptake) mode, and the potential impact of PTIP on transporter-based DDI risk prediction was assessed. The contribution of nonspecific binding and inhibitor toxicity to PTIP was interrogated through recovery and cell viability experiments. Correlations were sought between PTIP and such physicochemical descriptors of inhibitors as molecular weight, topological polar surface area (tPSA), cLogP, LogD7.4, and passive permeability (Papp) to establish if any of these parameters is predictive of the PTIP behavior of a drug. Finally, cellular accumulation and inhibitory potency of selected compounds were monitored over time to see whether the extent of PTIP correlates with intracellular unbound inhibitor concentration.
Materials and Methods
Transporter Cell Lines.
Human embryonic kidney 293 (HEK293) cell lines overexpressing the human uptake transporters OATP1B1, OATP1B3, OAT1, OAT3, OCT1, and OCT2, as well as Madin-Darby canine kidney II (MDCKII) cell lines overexpressing MATE1 and MATE2-K were created by lentiviral transduction. cDNAs were synthesized and cloned by GenScript (Piscataway, NJ), and lentiviral particles were generated in HEK293FT cells (Invitrogen/ThermoFisher, Waltham, MA). Transduced and antibiotic-selected cells were subjected to single-cell cloning, and amplified clones were functionally tested for transporter-specific uptake activity and stability of expression. Cell cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM), 4500 mg/l of glucose, supplemented with GlutaMAX, 10% v/v fetal bovine serum (FBS) and 1% v/v penicillin-streptomycin (all from Gibco/ThermoFisher).
Test Compounds.
Thirty transport inhibitors (Table 1) were selected to represent a broad range of molecular properties, including inhibitory potencies (Drug Interaction Database, University of Washington), physicochemical characteristics, and MDCK (low efflux) permeability (for measurement and calculation methods see Physicochemical Parameters of Test Compounds). Atorvastatin and pravastatin were purchased from Sequoia Research Products (Pangbourne, UK); daclatasvir, dolutegravir, isavuconazole, and ledipasvir were purchased from MedChem Express (Sollentuna, Sweden); ranitidine was purchased from abcr (Zug, Switzerland); all other inhibitors were purchased from Sigma-Aldrich (St. Louis, MO).
Physicochemical parameters vs. highest confirmed PTIP values of all test compounds used
Molecular properties were measured (LogD7.4, MDCK-LE Papp) or calculated (tPSA, cLogP, Kp,pred) as described in Materials and Methods. Highest PTIP values for each inhibitor are extracted from Table 2; where available, PTIP determined under “NSB block +” conditions was considered as confirmed. The respective transporter is noted in brackets. PTIP values ≥2.5x are highlighted in bold.
Physicochemical Parameters of Test Compounds.
For the determination of apparent Papp, MDCK (low efflux) cells (MDCK-LE, cat. no. 8412903, selected clone with low efflux activity; Sigma) were seeded on Transwell 96-well plate inserts (Corning, Tewksbury, MA) at a density of 2.6 × 105 cells/cm2 in high glucose DMEM with GlutaMAX containing 10% v/v FBS and 1% v/v penicillin-streptomycin (all from Gibco) and grown for 4 days at 37°C in an atmosphere of 5% CO2 and 95% relative humidity. Stock solutions of the compounds were prepared in dimethyl sulfoxide (DMSO) (10 mM), and each compound was dosed in triplicate at a final concentration of 10 μM in Hanks’ balanced salt solution (HBSS) at pH 7.4 containing 10 mM HEPES and 0.02% w/v bovine serum albumin (BSA). Cells were incubated with the compounds for 2 hours at 37°C, and flux was measured in the apical-to-basolateral direction. Dosing solutions as well as the calibration solutions were centrifuged for 10 minutes at 1000g. Drug concentrations in the donor and acceptor compartments were measured by liquid chromatography–mass spectrometry (LC-MS), as described in LC-MS Sample Preparation and Analytics, using a six-point calibration curve, glyburide as an analytical internal standard, and the settings listed in Supplemental Table 1.
LogD7.4 values were determined as described by Low et al. (2016). Topological polar surface area was calculated as described by Ertl et al. (2000). ClogP values were computed using CLOGP Daylight Version 4.9 (http://www.daylight.com/dayhtml/doc/clogp/; BioByte, Claremont, CA). Cellular Kp was predicted using the method described by Yabe et al. (2011) on the basis of logD7.4:
 (1)
(1)Investigation of Nonspecific Binding.
A subset of test compounds was investigated for nonspecific binding to plasticware. Polypropylene deep-well plates were precoated for 2 hours at 37°C with 200 μl/well HBSS buffer containing 20% v/v FBS and 2% w/v BSA. The coating solution was aspirated, and the plates were dried by nitrogen flow. Dosing solutions containing CsA, saquinavir, venetoclax, and ledipasvir were prepared at 0.1 and 1 μM in protein-free DMEM medium and dispensed into noncoated or precoated plates. Recovery after 0, 15, 30, 60, 120, and 180 minutes of incubation was measured by LC-MS as described below.
Uptake Inhibition Assays with Transporter Cell Lines.
Transporter-expressing cells and corresponding mock-transduced controls were seeded on untreated (if MDCKII-derived) or poly-d-lysine-treated (if HEK293-derived) 96-well tissue culture plates at a density of 1 × 105 cells/well. Sixteen to twenty hours after seeding, cells were rinsed twice with serum-free medium (SFM), then preincubated for 3 hours in the CO2 incubator with 1) SFM + DMSO or 2) dosing solutions containing serial dilutions of the test compound in SFM. For all conditions, the final DMSO concentration was adjusted to 1% v/v.
Following preincubation, cells were rinsed twice with assay buffer, and coincubation was started with a suitable radiolabeled probe and the dilution series of the inhibitor. A reference inhibitor instead of the test inhibitor was added to control wells to determine uptake in the absence of active transport. Assay conditions and specific reagents are summarized in Supplemental Table 2. Uptake was terminated by rinsing twice with ice-cold assay buffer, and cells were lysed with 0.1 M NaOH. Radioactivity in cell lysates mixed with liquid scintillation cocktail (Ultima Gold XR; PerkinElmer, Waltham, MA) was measured with a MicroBeta2 microplate counter (PerkinElmer).
In a first series of experiments, dosing solutions and assay reaction mixtures were kept in untreated 96-well cell culture plates (called auxiliary plates) for the duration of preincubation. In a second series, auxiliary plates were precoated as described in Investigation of Nonspecific Binding.
Determination of IC50 and PTIP Values.
Percent of control transport values were plotted against the logarithm of test inhibitor concentration, and IC50 values were calculated using a Log(inhibitor) versus normalized-response nonlinear regression model according to eq. 2, where HillSlope is a dimensionless curve fitting parameter: (2)PTIP was defined as the fold difference between IC50 values obtained without versus with a 3-hour preincubation. PTIP values ≥2.5 (i.e., preincubation caused a 2.5-fold or greater decrease in IC50) were considered potentially relevant, and transporter/test inhibitor pairs with PTIP ≥2.5 were taken forward for further investigation.
(2)PTIP was defined as the fold difference between IC50 values obtained without versus with a 3-hour preincubation. PTIP values ≥2.5 (i.e., preincubation caused a 2.5-fold or greater decrease in IC50) were considered potentially relevant, and transporter/test inhibitor pairs with PTIP ≥2.5 were taken forward for further investigation.
Cell Viability Assays and Estimation of Toxicity-Related IC50 Change.
In single-point toxicity assays, cell viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) according to the manufacturer’s instructions. Compounds that caused ≥10% loss of viability upon 3 hours of incubation were further tested in serial dilutions, and cell viability after 3 hours of treatment was measured by resazurin assay. Briefly, following the treatment cells were rinsed and incubated for additional 1 hour with 70 μM resazurin in SFM. Resorufin formation was quantified at 544 nm (ex)/620 nm (em) using a FluoStar Omega microplate reader (BMG Labtech, Ortenberg, Germany). The potential effect of toxicity on IC50 was estimated by multiplying percent transport values measured in the no-preincubation setting with percent viability at each compound concentration. An inhibition curve was fitted on the viability-modulated percent transport values according to eq. 2, IC50 was computed, and the fold change in IC50 potentially caused by toxicity was calculated.
DDI Risk Prediction on the Basis of In Vitro Results.
Guidelines issued by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) recommend the in vitro determination of IC50 or Ki for each novel drug candidate, and define formulas for the estimation of DDI risk on the basis of the relationship of IC50 or Ki to the unbound maximum plasma concentration in the systemic circulation or at the hepatic inlet (Supplemental Table 3). Owing to the uncertainties inherent to the calculation of maximum hepatic inlet concentration, we have chosen to estimate DDI risk for each transporter (including hepatic transporters) on the basis of Cmax,u,ss/IC50, where Cmax,u,ss was calculated as Cmax,ss, the maximum plasma concentration at steady state, multiplied by fu, the unbound fraction. Cmax,ss and fu were taken from the literature or the respective product labels (for references see Table 4), and fu values were replaced with 0.01 whenever the experimentally determined value was lower than 0.01.
Time Course of PTIP.
Transporter-expressing cells were preincubated for 15, 30, 60, 90, 120, and 180 minutes with test inhibitors previously shown to exhibit PTIP ≥2.5 upon 3 hours of preincubation. For each group, the total preincubation time of 180 minutes was split into a DMSO-only and an inhibitor interval (e.g., the 30-minute group was first incubated with DMSO for 150 minutes and then with inhibitor for 30 minutes). The control group was preincubated for 180 minutes with 1% v/v DMSO. After 180 minutes, an uptake assay was performed as described in Uptake Inhibition Assays with Transporter Cell Lines.
Time Course of Cellular Inhibitor Concentration.
Transporter-expressing cells were seeded at a density of 5 × 105 cells/well in poly-d-lysine-coated 24-well plates and grown for 16–18 hours. Before the experiment, a dosing solution (DS) containing the test compound was prepared in SFM, and the reference inhibitor of the transporter was added to an aliquot of the dosing solution (DS+Inh). An auxiliary 24-well plate used to contain DS and DS+Inh was precoated with 20% FBS/2% BSA to decrease nonspecific plastic binding of the test compound.
Treatment groups were incubated with DS for 1, 3, 5, 15, 30, 60, 90, 120, and 180 minutes, or with DS+Inh for 60 minutes and 180 minutes. The control group was incubated with SFM + DMSO for 180 minutes. Treatment groups incubated with DS+Inh, where the respective transporter was blocked, were included to assess the relative contribution of transporter-mediated uptake. For each group, the total preincubation time of 180 minutes was split into a DMSO-only and an inhibitor interval. Dosing solutions DS and DS+Inh were sampled at each dosing time point. At the endpoint (180 minutes), all supernatants were sampled, plates were placed on ice, cells were washed twice with ice-cold HBSS and extracted for 5 minutes with a 2:1 mixture of acetonitrile and deionized water. Collected samples of dosing solutions and supernatants were mixed 1:1 with acetonitrile.
LC-MS Sample Preparation and Analytics.
Cell extracts as well as dosing solution and supernatant samples were diluted as necessary and injected to LC-MS after addition of 0.5 μM glyburide as internal standard. The compounds were separated on a Phenomenex Synergi RP Polar column (2.5 μM, 30 × 2.1 mm; Phenomenex, Aschaffenburg, Germany) using water and acetonitrile including 0.1% formic acid (v/v) as mobile phases in gradient modes. The HPLC system was coupled to a QTRAP5500 mass spectrometer (AB Sciex LLC, Framingham, MA) using an electrospray interface. The mass spectrometer was operated in multiple reaction monitoring mode using the transitions shown in Supplemental Table 1. Data analysis was done with DiscoveryQuant and MultiQuant software (AB Sciex LLC).
Calculation of Cellular Uptake Parameters.
The cellular uptake was expressed as a clearance (PSinf; microliters per minute per 106 cells) calculated from the slope of the initial linear uptake phase normalized for the cell number (Vinf; picomoles per minute per 106 cells) and the applied concentration at time zero (C0; micromolars): (3)Kp describes the steady-state distribution of a compound between the cell and the medium and is calculated as the ratio of cellular and medium concentration at steady state (Ccell,ss/Cmedium,ss). When excluding active transport processes by the addition of an appropriate transporter inhibitor and assuming an unbound fraction (fu) of 1 in the protein-free medium, the unbound cellular fraction (fu,cell) can be derived as the reciprocal of Kp. Ccell,ss was calculated on the basis of the total cell number (5 × 105/well) and a cell volume of 0.8 μl/106 cells.
(3)Kp describes the steady-state distribution of a compound between the cell and the medium and is calculated as the ratio of cellular and medium concentration at steady state (Ccell,ss/Cmedium,ss). When excluding active transport processes by the addition of an appropriate transporter inhibitor and assuming an unbound fraction (fu) of 1 in the protein-free medium, the unbound cellular fraction (fu,cell) can be derived as the reciprocal of Kp. Ccell,ss was calculated on the basis of the total cell number (5 × 105/well) and a cell volume of 0.8 μl/106 cells. (4)Kp,uu is defined as the ratio of the unbound cellular concentration and the unbound medium concentration at steady state. Kp,uu can be calculated as the ratio of Kp values determined in the absence and presence of a reference inhibitor, assuming active transport was completely inhibited in the latter condition.
(4)Kp,uu is defined as the ratio of the unbound cellular concentration and the unbound medium concentration at steady state. Kp,uu can be calculated as the ratio of Kp values determined in the absence and presence of a reference inhibitor, assuming active transport was completely inhibited in the latter condition.
 (5)
(5)Statistical Analysis.
GraphPad Prism 7.03 (GraphPad Software, La Jolla, CA) was used to perform nonlinear regression, Receiver Operating Characteristic (ROC) analysis, as well as to calculate Spearman’s rank correlations. PTIP-positive or PTIP-negative status was assigned to each inhibitor depending on whether it displayed PTIP ≥2.5 in any assay, and ROC analysis was used to find cut-off values of physicochemical parameters that predict PTIP status with high sensitivity and specificity. Fisher’s exact test of contingency tables was done using VassarStats (http://vassarstats.net/). P values <0.05 were considered as significant.
Results
Occurrence of PTIP among Inhibitors of OATPs, OATs, OCTs, and MATEs.
Uptake inhibition assays were performed using HEK-OATP1B1, HEK-OATP1B3, HEK-OAT1, HEK-OAT3, HEK-OCT1, HEK-OCT2, MDCKII-MATE1, and MDCKII-MATE2-K cell lines with or without a 3-hour preincubation step. Since the phenomenon of PTIP has been most extensively studied on OATPs, only six compounds including the model time-dependent inhibitor CsA were examined in this set of studies. PTIP mediated by OATs, OCTs, and MATEs was examined with 10, 14, and 8 inhibitors, respectively. Seventy-six transporter/inhibitor combinations were analyzed in total. On the basis of the analysis of all PTIP values, including expected positives and negatives, the threshold for positive PTIP was set to 2.5-fold difference between IC50 values obtained without versus with preincubation.
Initially, dosing solutions and assay reaction mixtures were prepared in untreated 96-well cell culture plates (called auxiliary plates) and kept there for the duration of preincubation. Subsequently, assays with a positive PTIP result were repeated (N = 2) using auxiliary plates precoated with 20% v/v FBS and 2% w/v BSA. This surface treatment was shown to effectively reduce time-dependent nonspecific binding of four notoriously plastic-adherent compounds, saquinavir, CsA, venetoclax, and ledipasvir (Supplemental Fig. 1).
PTIP values from the initial series of experiments performed in uncoated plates (“NSB block −”) as well as from confirmatory experiments using precoated plates (“NSB block +”) are shown in Table 2. All corresponding IC50 values are listed in Supplemental Table 4. In the “NSB block −” experiments, three out of six OATP inhibitors, 1 out of 10 OAT inhibitors, 9 out of 14 OCT inhibitors, and 2 out of 8 MATE inhibitors were found to exhibit PTIP on at least one member of the respective transporter family. The most pronounced preincubation effect was observed with ledipasvir on HEK-OCT1 cells and venetoclax on HEK-OATP1B1 cells (Fig. 1, A and D). Ledipasvir had a similar, albeit smaller, effect on OCT2 transport activity when this transporter was expressed in HEK293 or MDCKII cells, suggesting that the inhibition specifically targets OCT activity and is not an artifact owing to the use of a particular host cell line (Fig. 1, B and C). Some inhibitors showed PTIP on multiple transporter families (saquinavir on OATP1Bs and OCT1; isavuconazole and vandetanib on OCTs and MATE2-K). Taking these overlaps into account, 12 out of 30 (40%) inhibitors showed PTIP on at least one transporter.
The preincubation effect of selected inhibitors on OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2-K
IC50 was measured after a 3-h preincubation with vehicle only or inhibitor, and PTIP was calculated as the fold decrease in IC50 caused by preincubation. Experiments were performed using untreated plasticware (“NSB block −,” N = 1) or plasticware precoated against nonspecific binding (“NSB block +,” N = 2). In the case of replicate experiments, PTIP was calculated as the ratio of the means of N = 2 IC50 values corresponding to vehicle and inhibitor preincubation conditions. For raw IC50 data see Supplemental Table 4. PTIP values highlighted in bold are equal to or greater than the threshold of 2.5x.

The preincubation effect of ledipasvir (A, B, C) and venetoclax (D) on OCTs and OATP1B1, respectively. IC50 values without preincubation could not be determined owing to weak inhibition. Each measurement point represents N = 3 replicates from a single experiment.
Experiments repeated with precoated auxiliary plates verified the PTIP-positive status of 10 inhibitors. Initial positive results were not confirmed for benzbromarone and cetirizine. Additionally, isavuconazole turned out to be PTIP-negative on MATE2-K but remained PTIP-positive on both OCTs. Venetoclax/OATP1B1 and ledipasvir/OCT1 experiments were also repeated with 1% w/v BSA present throughout all steps including washes, preincubation, and coincubation. Although the addition of BSA changed the apparent IC50 values and reduced the difference between the no-preincubation and preincubation conditions, PTIP with BSA remained 25-fold higher or more for both compounds (Supplemental Fig. 2).
The Effect of PTIP-Positive Inhibitors on Cell Viability.
A 3-hour treatment with a single high dose of confirmed PTIP-positive inhibitors has indicated no toxicity (cell viability ≥90%) for CsA, daclatasvir, dolutegravir, irinotecan, or ledipasvir (Supplemental Table 5). Isavuconazole at 300 μM was selectively toxic to HEK-OCT2 cells, whereas saquinavir at 100 μM impaired the viability of HEK-OATP1B1 and HEK-OATP1B3 but not of HEK-OCT1. Venetoclax at 100 μM was moderately toxic to both HEK-OATP1B1 and HEK-OATP1B3, and verapamil at 100 μM mildly affected HEK-OCT1 viability. Vandetanib had the most severe effect on all treated cell lines as it caused complete cell detachment when applied at 300 μM.
Compounds that reduced cell viability by ≥10% at high doses were further tested in serial dilutions using the resazurin viability assay. Modeled inhibition curves reflecting the toxic effect of preincubation superimposed on transporter inhibition without preincubation were overlaid on the actual inhibition curves measured without and with preincubation (Supplemental Fig. 3), and corresponding IC50 values were calculated. Table 3 displays preincubation-related IC50 changes with and without correction for reduced viability. For inhibitors with no apparent toxicity, the latter value was postulated to be 1.0. Although the toxic effects of isavuconazole, vandetanib, and venetoclax were sufficient to alter IC50 measurements (toxicity caused an estimated 1.4-, 1.5-, and 4.8-fold decrease in IC50, respectively), all 10 inhibitors remained PTIP-positive after this correction.
The estimated contribution of toxicity to PTIP
To estimate fold IC50 change caused by the occasional toxicity of a test inhibitor, percent transport values measured without preincubation at each concentration of the compound were multiplied by percent viability values determined after a 3-h preincubation with the same concentration. Fold IC50 change owing to toxicity was either postulated to be 1.0 when the inhibitor did not impair viability by more than 10% even at the highest applied concentration, or estimated as (IC50 without toxicity)/(IC50 with toxicity). For more details please refer to the main text and Supplemental Fig.3.
The Effect of Assay Duration on the Magnitude of PTIP.
For PTIP-positive inhibitors (excluding venetoclax and ledipasvir for which no definite values for PTIP could be determined), the magnitude of PTIP generally decreased with longer assay incubation times. PTIP was, on average, 13.0 ± 12.7-fold in 1-minute assays (OCT2, N = 5), 4.28 ± 1.70-fold in 3-minute assays (OATP1B1/3, N = 4), 3.98 ± 1.30-fold in 5-minute assays (OCT1, N = 6), and 1.93 ± 0.74-fold in 15-minute assays (MATE1/2-K, N = 4). In particular, daclatasvir PTIP was 34.2 (OCT2) versus 5.65 (OCT1), isavuconazole PTIP was 13.2 (OCT2) versus 3.15 (OCT1) versus 1.53/1.59 (MATE1/2-K), and vandetanib PTIP was 4.19 (OCT2) versus 2.69 (OCT1) versus 1.81/3.00 (MATE1/2-K) (Fig. 2). In line with this notion, when OATP1B1/CsA inhibition assay was performed with no preincubation but for multiple assay duration times (1, 2, 5, 10, and 15 minutes), IC50 fell 7.91-fold from 0.878 μM at 1 minute to 0.111 μM at 15 minutes (Supplemental Fig. 4), suggesting that, depending on the kinetics of inhibition, an extended coincubation may leave less or no room for preincubation to make a difference.

PTIP and assay duration. On average, higher PTIP values were observed in shorter assays, and inhibitors applied in multiple assays (e.g., daclatasvir, isavuconazole) tended to display higher PTIP in assays with shorter coincubation time.
The Relevance of PTIP to In Vitro DDI Risk Assessment.
Using IC50 data of confirmed PTIP-positive clinical compounds from the second set of experiments (“NSB block +”), we projected the impact of preincubation on the outcome of DDI risk prediction. Cmax,u,ss/IC50 values were calculated from IC50 values without versus with preincubation and compared against cut-off values recommended by FDA, PMDA, and EMA (see Supplemental Table 3). For each drug, the outcome of in vitro DDI risk prediction was “risk” if Cmax,u,ss/IC50 exceeded the respective cut-off value in any of the assays performed with that compound, and “no risk” otherwise. As shown in Table 4, preincubation did not affect the outcome for CsA, daclatasvir, saquinavir, ledipasvir, or venetoclax. For dolutegravir, Cmax,u,ss/IC50 with preincubation exceeded the EMA cut-off but not the FDA/PMDA cut-off; thus, the outcome changed to “risk” according to the EMA criteria only. Finally, in the case of irinotecan, isavuconazole and vandetanib if only the recommended OCT2 assay was considered, preincubation changed the EMA outcome from “no risk” to “risk,” while the OCT1 and/or MATE assays (which are optional according to EMA guidelines) predicted “risk” even without preincubation. Hence, whether preincubation changed the EMA outcome for isavuconazole, irinotecan and vandetanib depended on the panel of assays considered.
The effect of preincubation on the outcome of in vitro DDI risk assessment
Maximum steady-state total plasma concentration (Cmax,ss) and unbound fraction (fu) values were taken from the sources referenced below the table. Maximum steady-state unbound plasma concentrations (Cmax,u,ss) were calculated as Cmax,ss * fu, and IC50 values are means from N = 2 “NSB block +” experiments (see Supplemental Table 4).
Correlation of PTIP with Physicochemical Descriptors of the Test Inhibitors.
Table 1 lists the physicochemical parameters and highest observed PTIP values (except for pravastatin for which no IC50 value could be determined) for all test compounds. Molecular weight (MW), tPSA, cLogP, LogD7.4, MDCK-LE Papp, Kp predicted from LogD7.4 (Kp, pred), as well as the ratio Kp, pred/MDCK-LE Papp were plotted against PTIP (Fig. 3). Spearman’s rank correlations were calculated between PTIP and each physicochemical descriptor.

Correlation of PTIP with physicochemical descriptors. PTIP showed significant nonparametric (Spearman’s) correlation with molecular weight (A), LogD7.4 (B), Kp,pred (C), and Kp,pred/MDCD-LE Papp (D), but not with tPSA (E), cLogP (F), or MDCK-LE Papp alone (G). Black dotted vertical lines indicate the PTIP threshold of 2.5; red dotted horizontal lines indicate the cut-off values determined by ROC analysis and used for the calculation of specificity/sensitivity. Instead of the indeterminate PTIP ranges of ledipasvir and venetoclax (>255 and >258) the respective minima were used (= 255 and = 258; red symbols).
Of the seven parameters considered, MW, LogD7.4, Kp, pred, and Kp, pred/MDCK-LE Papp showed significant correlation with PTIP (Spearman’s rho rSp = 0.68, 0.67, 0.67, and 0.58, respectively). On the other hand, tPSA, cLogP, and MDCK-LE Papp alone showed only weak or no correlation with PTIP. The descriptors were also found to be intercorrelated (for the correlation matrix see Supplemental Table 6; note that Kp, pred was calculated from LogD7.4, hence the rSp of 1.0).
To assess the predictive value of MW, LogD7.4, Kp, pred, and Kp, pred/MDCK-LE Papp with regard to PTIP status, ROC analysis was performed to find cut-off values with best sensitivity and specificity. MW >437 g/mol was the best single predictor of PTIP+ status (sensitivity: 0.90/specificity: 0.89, Fisher’s exact test: P = 0.00007), followed by LogD7.4 >1.85 or, equivalently, Kp, pred >24.85 (sensitivity: 0.90/specificity: 0.83, P = 0.00027, for both). Kp, pred/MDCK-LE Papp values greater than nine predicted positive PTIP with a sensitivity of 0.70 and a specificity of 0.89 (P = 0.0028).
Cellular Uptake of Inhibitors and PTIP: Time-Course Experiments.
Cellular uptake over time was investigated for the PTIP-positive inhibitors ledipasvir, venetoclax, CsA, and saquinavir, the PTIP-negative inhibitors valsartan and trimethoprim, as well as for benzbromarone that was PTIP-positive when using untreated auxiliary plates but turned negative upon plate precoating. Compounds were applied at concentrations close to the IC50 value with preincubation. Cellular uptake parameters are shown in Table 5; for comparability across PTIP-positive and PTIP-negative compounds, “NSB block −” PTIP values were used in calculations.
PTIP values and cellular uptake parameters of selected transporter/inhibitor pairs
For comparability across PTIP-positive and PTIP-negative transporter/inhibitor combinations, “NSB block −” PTIP values are shown in each case. When determined in the presence of a specific and potent inhibitor, Kp reflects passive equilibration. Kp,uu values >1 indicate active accumulation. For calculations see Materials and Methods.
Cell concentration time profiles are shown in Fig. 4 together with the degree of transport inhibition that was determined in independent experiments. Cellular concentrations did not reach steady-state over the 180-minute interval for ledipasvir and venetoclax, and for both compounds the degree of transport inhibition reached its maximum after a 120-minute preincubation. Time to cellular steady-state concentration was approx. 120 minutes for CsA and 60 minutes for saquinavir. With CsA, transport inhibition peaked after 90–120 minutes of preincubation; likewise, saquinavir reached maximum inhibition at 120 minutes. Cellular concentrations of benzbromarone reached steady-state after 60 minutes in OAT3 and 15 minutes in OAT1, but the degree of OAT3 inhibition was hardly affected by the duration of preincubation, which may explain the lack of observed PTIP. The PTIP-negative inhibitors valsartan and trimethoprim were identified as substrates of their respective transporters by Kp,uu values >1, and their cellular concentrations equilibrated rapidly with the medium (3 minutes and 1 minute to steady-state, respectively).

Cellular concentration and inhibition potency of selected inhibitors over time. Cellular concentration (green) was measured at multiple time points between 1 and 180 minutes; also, in the case of PTIP-positive inhibitors, inhibition potency (red) was determined at multiple preincubation durations ranging between 15 and 180 minutes. Dotted vertical lines indicate the time required to reach steady-state cellular concentration. Each point represents the mean ± S.D. of N = 3 replicates. For benzbromarone (OAT1), valsartan, and trimethoprim, no potency time profiles were recorded as no PTIP effect was observed, which is in line with steady-state concentrations being reached within a few minutes.
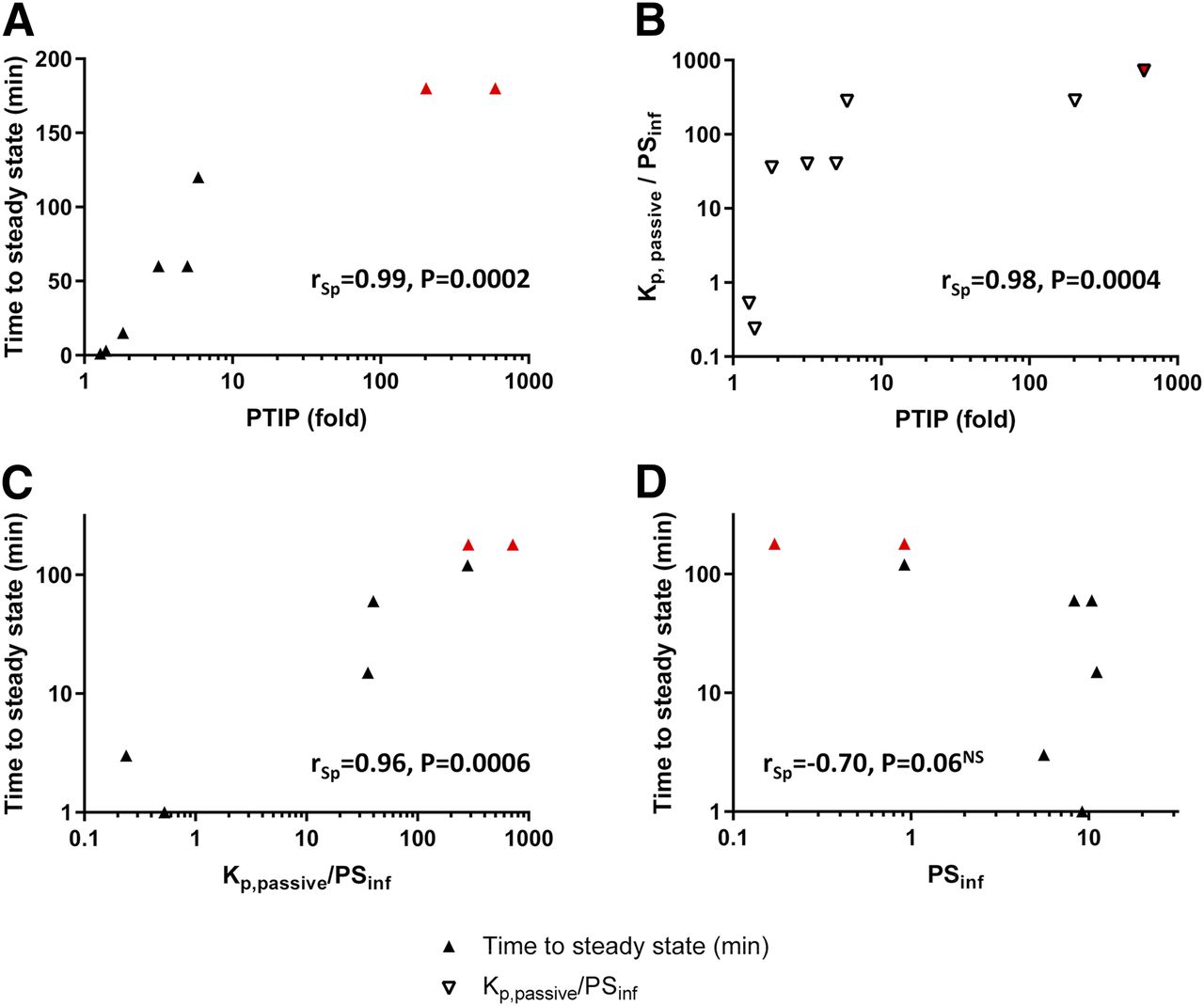
When looking for associations between 3-hour PTIP and cellular uptake parameters (Fig. 5), strong positive rank correlation was found between PTIP and time to steady-state (TTSS) (rSp = 0.99, P = 0.0002), as well as between PTIP and the ratio composed of passive cellular accumulation over initial influx, Kp,passive/PSinf (rSp = 0.98, P = 0.0004). Moreover, Kp,passive/PSinf and TTSS were positively intercorrelated (rSp = 0.96, P = 0.0006), whereas the negative correlation between PSinf alone and TTSS fell short of being significant (rSp = −0.70, P = 0.06).

Associations between PTIP and cellular uptake parameters. Correlations were calculated from data shown in Table 5. Positive association was found between PTIP and TTSS, as well as PTIP and Kp,passive/PSinf (A and B). The latter parameter was also positively correlated with TTSS (C), whereas no significant correlation was found between PSinf and TTSS (D). Instead of the indeterminate PTIP and/or TTSS ranges of ledipasvir and venetoclax, the respective minimum values are shown (red symbols). NS, not significant.
Discussion
Recent FDA guidance recommends a 30-minute preincubation when investigating the inhibitory potential of a test article against OATP1B1 and OATP1B3 (FDA, 2017). Little is known about the effect of preincubation on other transporters, or the molecular properties that predispose some compounds to PTIP.
Here we investigated the preincubation effect of 30 miscellaneous compounds on OATP1B1, OATP1B3, OAT1, OAT3, OCT1 OCT2, MATE1, and MATE2-K. A prolonged preincubation of 3 hours was chosen to allow for the equilibration of compounds with low permeability between the extracellular and intracellular compartments. Preincubation caused a shift toward lower IC50 values for the majority of compounds, and for 10 inhibitors the apparent potentiation was ≥2.5-fold (Table 2). A PTIP effect ≥2.5-fold was seen for half of OATP1B inhibitors and also more than half of OCT inhibitors, but not confirmed for OATs or MATEs.
A possible explanation for the higher frequency of PTIP among OCTs versus MATEs is the longer incubation time in MATE assays. PTIP ≥2.5-fold was mostly observed for OATP1Bs and OCTs with assay times between 1 and 5 minutes, whereas in MATE assays a 15-minute coincubation may have allowed inhibitors to equilibrate between the medium and intracellular space. The following findings support the notion that the preincubation effect is more pronounced when the subsequent coincubation is brief: 1) Assays with shorter incubation time were generally associated with higher PTIP values, 2) the PTIP effect of daclatasvir and isavuconazole was inversely related to assay duration (Fig. 2), and 3) prolonged coincubation markedly decreased IC50 of CsA on OATP1B1 (Supplemental Fig. 4).
This argument, however, fails to explain why no PTIP was observed for OAT assays with short incubation times (1 or 2 minutes). Although most investigated OAT inhibitors either had a comparatively high passive permeability (>3 × 10−6 cm/s) or were reported to be OAT substrates, bumetanide was also PTIP-negative despite a low MDCK-LE Papp (1.13 × 10−6 cm/s). The possible OAT-mediated bumetanide transport was not investigated in this study.
In general, we cannot exclude that other compounds not tested herein or the same compounds under different conditions may exhibit PTIP on OATs or MATEs. Published data on OAT-based preincubation effects exist (Ma et al., 2015), and the PTIP liability of MATEs cannot be dismissed solely on the basis of our results. Ultimately, it can also be speculated that some transporter families may be more prone to PTIP than others owing to inherent differences in protein structure or mechanism of transport.
Upon confirmation of the novel PTIP-positive inhibitors toward OATP1Bs and OCTs, additional investigations were done to verify the mechanism behind this phenomenon. Some PTIP-positive compounds were known to exhibit strong NSB, and the preincubation effect could have been mimicked by time-dependent adsorption of test compounds to the transport apparatus. Although this issue was not addressed in our first series of experiments, on repeating the PTIP-positive assays we used precoated plates in an attempt to minimize NSB. Reassuringly, the preincubation effect was confirmed in 19 out of 23 instances. Additionally, we re-examined the most distinct cases of PTIP (venetoclax/OATP1B1 and ledipasvir/OCT1) in the presence of 1% w/v BSA that was expected to limit the nonspecific binding (Supplemental Fig. 2). As PTIP was not eliminated by the addition of BSA, a major role of NSB in the preincubation effect seemed improbable.
Cytotoxicity of the test compounds was considered as another potential confounding factor. Toxicity of an inhibitor is barely of concern in a conventional assay containing a short coincubation only, but a 3-hour preincubation with the same compound may severely compromise cell viability and/or attachment. Unless substrate uptake is normalized for total cellular protein content, cell loss or impaired viability may be mistaken for transport inhibition. Five out of ten PTIP-positive compounds caused marked cell death; however, toxic concentration ranges were typically 10- to 100-fold higher than the respective IC50 values. Thus, except for a single case (venetoclax on HEK-OATP1B3), toxicity had minor or no contribution to the preincubation effect.
Taken together, these experiments highlighted OCTs as a new family of uptake transporters sensitive to preincubation and confirmed PTIP as a true effect not caused by NSB and/or toxicity. Experiments proving ledipasvir PTIP in MDCKII-OCT2 cells (Fig. 1) raised confidence that our findings were not exclusive to HEK293 cells.
Importantly, by calculating Cmax,u,ss/IC50 values without and with preincubation we have shown that preincubation may alter the outcome of in vitro DDI risk assessment. In our experiments, preincubation changed the DDI prediction from “no risk” to “risk” for dolutegravir, isavuconazole, irinotecan, and vandetanib according to the EMA guidelines. Clinical data have confirmed vandetanib and dolutegravir as perpetrators of transporter-based DDI (Reese et al., 2013; Johansson et al., 2014). Of note, preincubation did not change the predicted DDI risk of ledipasvir and venetoclax despite their robust PTIP effects. This is because both drugs have extremely low measured plasma fu values (<0.002 for ledipasvir, 0.000013 for venetoclax) (https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205834s000lbl.pdf; Emami Riedmaier et al., 2018). Even if fu is set to 0.01 as a “worst-case” assessment, the Cmax,u,ss/IC50 ratio remains below the cut-off.
To elucidate the possible mechanism of the preincubation effect we measured the time profiles of cellular concentration and transport inhibition. For ledipasvir, venetoclax, and CsA, cellular concentration and inhibitory potency followed remarkably similar time courses (Fig. 4). Although the measured PSinf values did not correlate significantly with TTSS, the correlation could be greatly improved by dividing Kp,passive by PSinf (Fig. 5). The latter describes the steady-state passive distribution of a compound between the cells and the medium, and in a protein-free medium high Kp,passive values indicate substantial cellular binding. Low passive permeation combined with high cellular binding results in a slow build-up of intracellular unbound drug concentration. Our data support a conclusion that intracellular unbound perpetrator concentration is a major determinant of inhibitory potency, and, consequently, the magnitude of PTIP depends on the equilibration time.
To explore whether PTIP can be predicted from a priori physicochemical parameters, we correlated PTIP with MDCK-LE permeability and molecular properties. Unlike tPSA, cLogP, or MDCK-LE Papp, physicochemical descriptors including MW, LogD7.4- and the LogD7.4-derived Kp,pred, as well as Kp,pred/MDCK-LE correlated with the preincubation effect (Fig. 3). High (>ca. 440 Da) MW was the most reliable predictor of PTIP-positive behavior. Although the data plots were suggestive of a linear relationship between Log(PTIP) and Kp,pred, as well as Log(PTIP) and Kp,pred/MDCK-LE Papp, these data series were skewed by the few high-PTIP inhibitors such as venetoclax and ledipasvir. Therefore, an attempt to quantitatively predict PTIP on the basis of these two parameters would be too ambitious until more data has been collected.
It remains to be seen whether in vitro assays with preincubation produce more physiologically relevant data for physiology-based pharmacokinetic (PBPK) models. Underestimated inhibitory potency is among the many reasons why in vitro data-fed PBPK models perform poorly at predicting transporter-based DDI (Jamei et al., 2014; Pan et al., 2016). Preincubation improved the predictivity of a PBPK model describing the interaction of CsA with OATP1B1 (Shitara and Sugiyama, 2017). It will also need to be tested if preincubation can help decrease high interlaboratory variation in transporter inhibition data. Reported IC50 or Ki values of OATP1B1 inhibitors sometimes vary across several orders of magnitude (Vaidyanathan et al., 2016). Here we also showed that without preincubation, extending assay duration from 1 to 15 minutes caused an almost 8-fold decrease in IC50 (Supplemental Fig. 4).
In summary, we demonstrate that PTIP in vitro affects transport activity of OCTs as well as of OATP1Bs. Our data demonstrate that the inclusion of appropriate control experiments is crucial before concluding that true PTIP was observed. Most notably, control experiments to exclude effects of nonspecific binding and cellular toxicity as confounding factors should be performed. Since PTIP results in reduced IC50 values, the number of positive in vitro DDI risk predictions is generally expected to increase, unless I/IC50 cut-off values in guidelines are adjusted accordingly. The propensity of an inhibitor for PTIP depends on molecular properties such as high molecular weight and hydrophobicity, and a slow build-up of the unbound intracellular perpetrator drug concentration is a prerequisite for the preincubation effect.
Acknowledgments
The authors acknowledge John P. Keogh (JPK Consulting, Hitchin, United Kingdom) for his invaluable help with the initiation of the project and suggestions on the study design, and Stephane Rodde (Novartis Pharma AG) for measuring LogD7.4 values.
Authorship Contributions
Participated in research design: Tátrai, Schweigler, Poller, Domange, Hanna, de Wilde, Gáborik, Huth
Conducted experiments: Tátrai, Domange.
Contributed new reagents or analytic tools: Poller, Domange, Huth.
Performed data analysis: Tátrai, Schweigler, Poller, Domange, Gáborik, Huth.
Wrote or contributed to the writing of the manuscript: Tátrai, Schweigler, Poller, Domange, Hanna, de Wilde, Gáborik, Huth.
Footnotes
- Received January 8, 2019.
- Accepted May 2, 2019.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BSA
- bovine serum albumin
- CsA
- cyclosporin A
- DDI
- drug-drug interaction
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethyl sulfoxide
- DS
- dosing solution
- EMA
- European Medicines Agency
- FBS
- fetal bovine serum
- FDA
- Food and Drug Administration
- HBSS
- Hanks’ balanced salt solution
- HEK
- human embryonic kidney
- Inh
- inhibitor
- LC-MS
- liquid chromatography–mass spectrometry
- MATE
- multidrug and toxin extrusion
- MDCK
- Madin-Darby canine kidney
- MDCK-LE
- MDCK-low efflux
- MW
- molecular weight
- NSB
- nonspecific binding
- OAT
- organic anion transporter
- OATP
- organic anion transporting polypeptide
- OCT
- organic cation transporter
- Papp
- passive permeability
- PBPK
- physiology-based pharmacokinetic
- PMDA
- Pharmaceuticals and Medical Devices Agency
- PTIP
- potentiation of transporter inhibition by preincubation
- ROC
- receiver operating characteristic
- SFM
- serum-free medium
- tPSA
- topological polar surface area
- TTSS
- time to steady state
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}