


Abstract
The glucose-lowering drug pioglitazone undergoes hepatic CYP2C8-mediated biotransformation to its main metabolites. The antiplatelet drug clopidogrel is metabolized to clopidogrel acyl-β-d-glucuronide, which was recently found to be a strong time-dependent inhibitor of CYP2C8 in humans. Therefore, we studied the effect of clopidogrel on the pharmacokinetics of pioglitazone. In a randomized crossover study, 10 healthy volunteers ingested either 300 mg of clopidogrel on day 1, and 75 mg on days 2 and 3, or placebo. Pioglitazone 15 mg was administered 1 hour after placebo and clopidogrel on day 1. Plasma concentrations of pioglitazone, clopidogrel, and their main metabolites were measured up to 72 hours. Clopidogrel increased the area under the plasma concentration-time curve (AUC0–∞) of pioglitazone 2.1-fold [P < 0.001, 90% confidence interval (CI) 1.8–2.6] and prolonged its half-life from 6.7 to 11 hours (P = 0.002). The peak concentration of pioglitazone was unaffected but the concentration at 24 hours was increased 4.5-fold (range 1.6–9.8; P < 0.001, 90% CI 3.17–6.45) by clopidogrel. The M-IV-to-pioglitazone AUC0–∞ ratio was 49% (P < 0.001, 90% CI 0.40–0.59) of that during the control phase, indicating that clopidogrel inhibited the CYP2C8-mediated biotransformation of pioglitazone. Clopidogrel increases the exposure to pioglitazone by inhibiting its CYP2C8-mediated biotransformation. In consequence, use of clopidogrel may increase the risk of fluid retention and other concentration-related adverse effects of pioglitazone.
Introduction
The thiazolidinedione pioglitazone is an oral glucose-lowering drug that acts by activating the peroxisome proliferator-activated receptor γ (PPAR-γ) (Diamant and Heine, 2003; Waugh et al., 2006). Pioglitazone is well absorbed, has an oral bioavailability of about 80%, and is extensively metabolized to active and inactive metabolites in the liver (Eckland and Danhof, 2000). The main metabolites found in human plasma, hydroxypioglitazone (M-IV), and its secondary metabolite, ketopioglitazone (M-III), contribute to the pharmacological activity (Eckland and Danhof, 2000). Pioglitazone is metabolized mainly by the cytochrome P450 CYP2C8 and to a minor extent by CYP3A4 (Jaakkola et al., 2006a). Certain drugs and genetic factors can have a clinically significant effect on hepatic CYP2C8 activity (Backman et al., 2016), and therefore such factors can alter the exposure to pioglitazone (Deng et al., 2005; Jaakkola et al., 2005, 2006b; Tornio et al., 2008; Aquilante et al., 2013b; Kadam et al., 2013), potentially changing its efficacy and the risk for concentration-related adverse effects. For example, the fibric acid derivative gemfibrozil, which is a strong CYP2C8 inhibitor, raises the exposure to pioglitazone more than 3-fold (Deng et al., 2005; Jaakkola et al., 2005; Aquilante et al., 2013b), and the product information of Actos warns about risks of their combination (European Medicines Agency, 2014).
Antithrombotic agents, such as the ADP-receptor inhibitor clopidogrel, are commonly used in patients with type 2 diabetes mellitus. In a retrospective epidemiologic study, concomitant use of clopidogrel was strongly associated with myotoxic adverse effects of cerivastatin, which is a substrate for CYP2C8 (Mück, 2000; Backman et al., 2002; Floyd et al., 2012). More recently, therapeutic doses of clopidogrel were shown to markedly increase the plasma concentrations and blood glucose-lowering effect of the meglitinide analog repaglinide (Tornio et al., 2014). Inhibition of CYP2C8 by clopidogrel metabolite clopidogrel acyl-β-d-glucuronide was suggested to be the main mechanism of the clopidogrel-repaglinide interaction. Therefore, and as the metabolism of pioglitazone is mainly CYP2C8-mediated (Gillies and Dunn, 2000; Jaakkola et al., 2006a; Scheen, 2007; Tornio et al., 2012), we found it important to investigate the effect of clopidogrel on the pharmacokinetics of pioglitazone.
Materials and Methods
Subjects and Study Design.
Ten healthy nonsmoking volunteers (4 women, 6 men; age range, 20–35 years; body mass index range, 19.2–28.9 kg/m2) participated in the study after giving written informed consent. Their health was confirmed by medical history, clinical examination, and routine laboratory tests before they entered the study. All participants had normal blood platelet counts and hemoglobin values. None of the subjects used oral contraceptives or other continuous medication. The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (record number 259/13/03/00/2014), and the Finnish Medicines Agency Fimea (EudraCT number 2013-003891-11). In a randomized, placebo-controlled, crossover study, the subjects ingested as pretreatment either placebo (placebo tablets; University Pharmacy, Helsinki, Finland) or clopidogrel (Plavix; Sanofi-Aventis, Paris, France) once daily at 8 AM for 3 days with 150 ml water. Clopidogrel dose was 300 mg (four 75-mg tablets) on day 1 followed by 75 mg on days 2 and 3. The wash-out period between the placebo and clopidogrel phases was 3 weeks. On day 1 of pretreatment, 15 mg of pioglitazone (Actos 15 mg tablet; Takeda Europe, London, UK) was administered at 9 AM. On the days of pioglitazone intake, the volunteers had fasted overnight, and a standard warm meal was served 3 hours and snacks 7 and 10 hours after the administration of pioglitazone. The use of grapefruit products was not allowed for 1 week before and during the study, and the use of other drugs from 1 week before to 1 week after the study. Use of alcohol was prohibited the day before and on the days of pioglitazone administration.
Sampling.
Timed blood samples were drawn from a cannulated forearm vein, or by venipuncture before the administration of pretreatment, and 5 minutes before and 1, 2, 3, 4, 5, 7, 9, 12, 24, 48, and 72 hours after the administration of pioglitazone. The blood samples were collected into EDTA-containing tubes, which were placed on ice immediately after sampling. Plasma was separated within 30 minutes and stored at –70°C until analysis. The samples for the determination of clopidogrel and its metabolites were treated with 2-bromo-3′-methoxyacetophenone within 30 seconds of blood sample collection in whole blood EDTA samples to derivatize the active metabolite of clopidogrel, as previously described (Delavenne et al., 2010).
Determination of Drug Concentrations.
The concentrations of pioglitazone, M-III, and M-IV were measured with an Agilent 1100 HPLC system (Agilent Technologies, Waldbronn, Germany) coupled to an AB Sciex API 2000 tandem mass spectrometer (Framingham, MA), as previously described, with slight modifications (Jaakkola et al., 2005; Kawaguchi-Suzuki et al., 2014). Briefly, the plasma (100 μl) proteins were precipitated with a volume of 300 μl of acetonitrile containing 1% formic acid and the internal standards, and the supernatant phospholipids were removed using HybridSPE Phospholipid cartridges (30 mg; Supelco/Sigma-Aldrich, Bellefonte, PA). The resulting filtrate was evaporated and the residue was reconstituted in 80 μl of mobile phase, and injected (5 μl) into the liquid chromatography instrument. Chromatography was performed on XTerra RP C18 column (3.9 × 100 mm; Waters Corporation, Milford, MA) using gradient elution. The mobile phase consisted of 10 mM ammonium acetate (pH 9.0, adjusted with 25% ammonia solution) and acetonitrile. Deuterated forms of pioglitazone, M-III, and M-IV served as internal standards. The mass spectrometer was operated in positive electrospray mode, and the mass-to-charge (m/z) ion transitions [M+H]+ monitored were 357–134 for pioglitazone, 371–148 for M-III, and 373–150 for M-IV. The limit of quantification was 0.2 ng/ml for pioglitazone and M-III, and 0.5 ng/ml for M-IV. The day-to-day coefficient of variation (CV) was below 15% for all analytes at relevant concentrations. Clopidogrel, the clopidogrel active cis-5-thiol metabolite, clopidogrel carboxylic acid, and clopidogrel acyl-β-d-glucuronide were analyzed with a Nexera UPLC instrument (Shimadzu, Kyoto, Japan) connected to an AB Sciex QTrap 5500 mass spectrometer, as previously described (Holmberg et al., 2014; Tornio et al., 2014; Itkonen et al., 2015).
Pharmacokinetics.
The peak plasma concentration (Cmax), time to Cmax (tmax), area under the concentration-time curve (AUC)0–72h, area under the plasma concentration time curve from zero to infinity (AUC0–∞), and half-life (t1/2) were calculated for pioglitazone, clopidogrel, and their metabolites (AUC0–13h for clopidogrel and its metabolites) by standard noncompartmental methods using Phoenix WinNonlin, version 6.4 (Certara, Princeton, NJ).
Static In Vitro–In Vivo Predictions of the Clopidogrel-Pioglitazone Interaction.
Previously published inhibitor concentration that supports half-maximal rate of inactivation (KI), and maximal inactivation rate (kinact) values of clopidogrel acyl-β-d-glucuronide for CYP2C8 were used to predict the in vivo clopidogrel-pioglitazone interaction with the following equation (Mayhew et al., 2000; Jones and Hall, 2002; Tornio et al., 2014):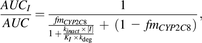 where AUCI and AUC are the areas under the concentration–time curve of pioglitazone with clopidogrel (inhibitor) and placebo (control), respectively; fmCYP2C8 expresses the fraction of pioglitazone dose metabolized by CYP2C8; and kdeg is the rate constant of hepatic CYP2C8 degradation in the absence of the inhibitor. In our predictions, a half-life of 22 hours was used for CYP2C8, corresponding to a kdeg of 0.00053 1/min (Backman et al., 2009). The predictions were calculated for clopidogrel acyl-β-d-glucuronide concentrations ranging from 0 to 10 μM and for fmCYP2C8 values of 0.60, 0.65, 0.75, and 0.80. The predictions were then related to the median unbound plasma peak (Cmax,u) and average (Cavg,u) concentrations of clopidogrel observed in the clinical study, assuming an unbound fraction of 10% (Tornio et al., 2014), where the average concentration was calculated by dividing AUC0–13h by 13 hours. Clopidogrel and its other metabolites were not included in the predictions because of their negligible significance (Tornio et al., 2014).
where AUCI and AUC are the areas under the concentration–time curve of pioglitazone with clopidogrel (inhibitor) and placebo (control), respectively; fmCYP2C8 expresses the fraction of pioglitazone dose metabolized by CYP2C8; and kdeg is the rate constant of hepatic CYP2C8 degradation in the absence of the inhibitor. In our predictions, a half-life of 22 hours was used for CYP2C8, corresponding to a kdeg of 0.00053 1/min (Backman et al., 2009). The predictions were calculated for clopidogrel acyl-β-d-glucuronide concentrations ranging from 0 to 10 μM and for fmCYP2C8 values of 0.60, 0.65, 0.75, and 0.80. The predictions were then related to the median unbound plasma peak (Cmax,u) and average (Cavg,u) concentrations of clopidogrel observed in the clinical study, assuming an unbound fraction of 10% (Tornio et al., 2014), where the average concentration was calculated by dividing AUC0–13h by 13 hours. Clopidogrel and its other metabolites were not included in the predictions because of their negligible significance (Tornio et al., 2014).
Genotyping.
Buffy coats were prepared from 9 ml whole blood EDTA samples after plasma separation. Genomic DNA was extracted from the buffy coats using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). The participants were genotyped for the CYP2C8*2 (c.805A>T), *3 (c.416G>A and c.1196A>G), and *4 (c.792C>G) alleles with a custom TaqMan OpenArray genotyping panel on an Applied Biosystems QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Haplotypes containing the reference allele at each position were designated as CYP2C8*1.
Statistical Analysis.
On the basis of the pharmacokinetic results of previous drug interaction studies with pioglitazone, 10 subjects were estimated to be adequate to detect a 30% change in the AUC of pioglitazone and its active metabolites between the control and clopidogrel phases, with a power of at least 80% (risk of 5% for type I error). The results are expressed as geometric means and geometric mean ratios with geometric CV or 90% confidence intervals (CIs) unless stated otherwise. Logarithmic transformation was used for pharmacokinetic variables, except tmax, before statistical analysis. The pharmacokinetic variables were compared by repeated-measures analysis of variance with treatment phase as a within-subjects and treatment sequence as a between-subjects factor, with pairwise comparisons using Fisher’s least significant difference method. The tmax data were compared using the Wilcoxon signed rank test. Correlations between the AUC0–13h of clopidogrel, or its metabolites, and the fold-increase in pioglitazone AUC0–∞ were quantified as Kendall’s correlation coefficients and tested using Kendall’s test. The M-IV-to-pioglitazone metabolic ratio and pioglitazone AUC0–∞ of CYP2C8*3 carriers were compared with those of CYP2C8*3 noncarriers by independent-samples t tests. The P-values below 0.05 were considered statistically significant. Statistical analyses were performed using SPSS Statistics for Windows version 22.0 (IBM Corporation, Armonk, NY).
Results
Pioglitazone.
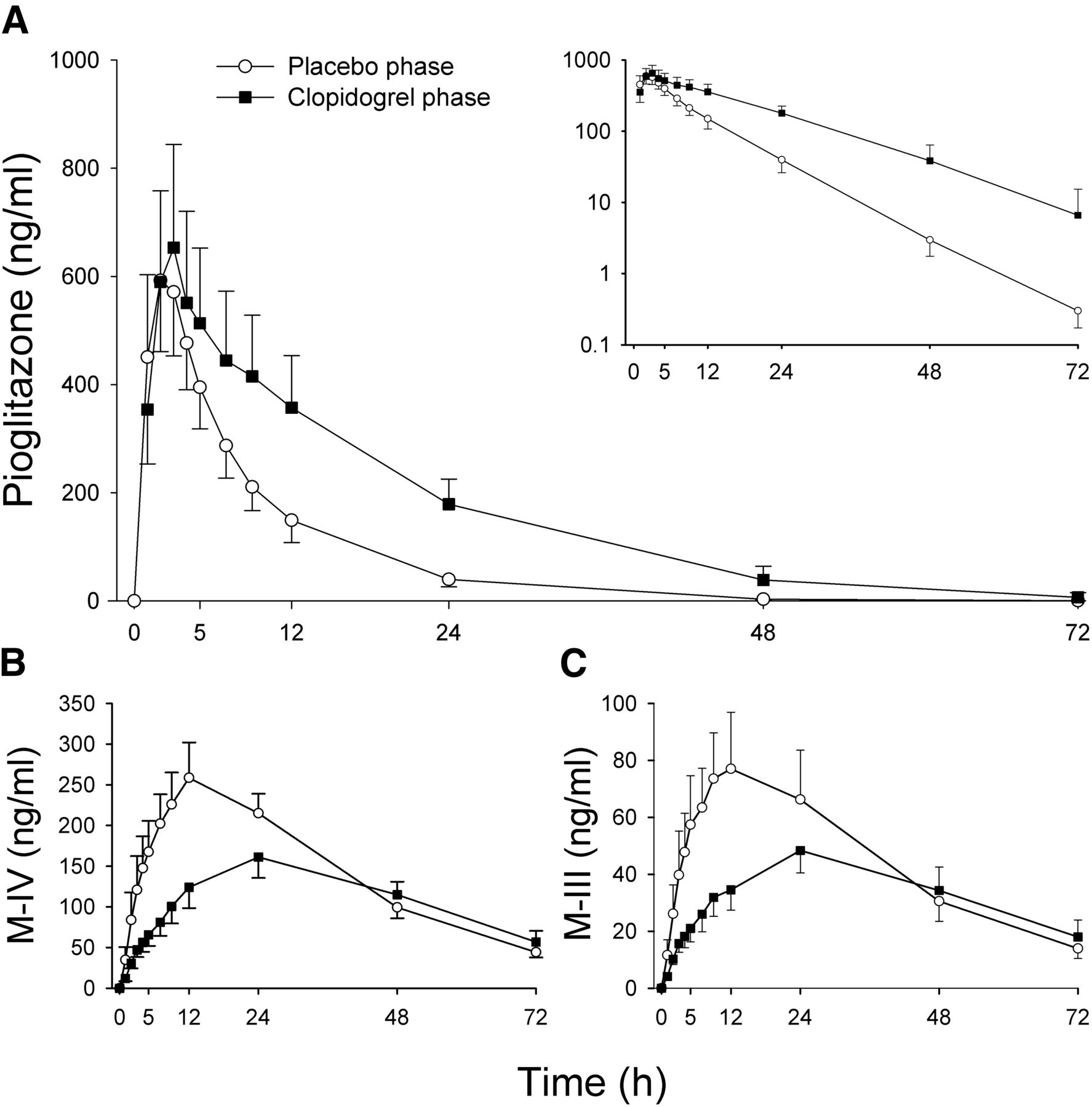
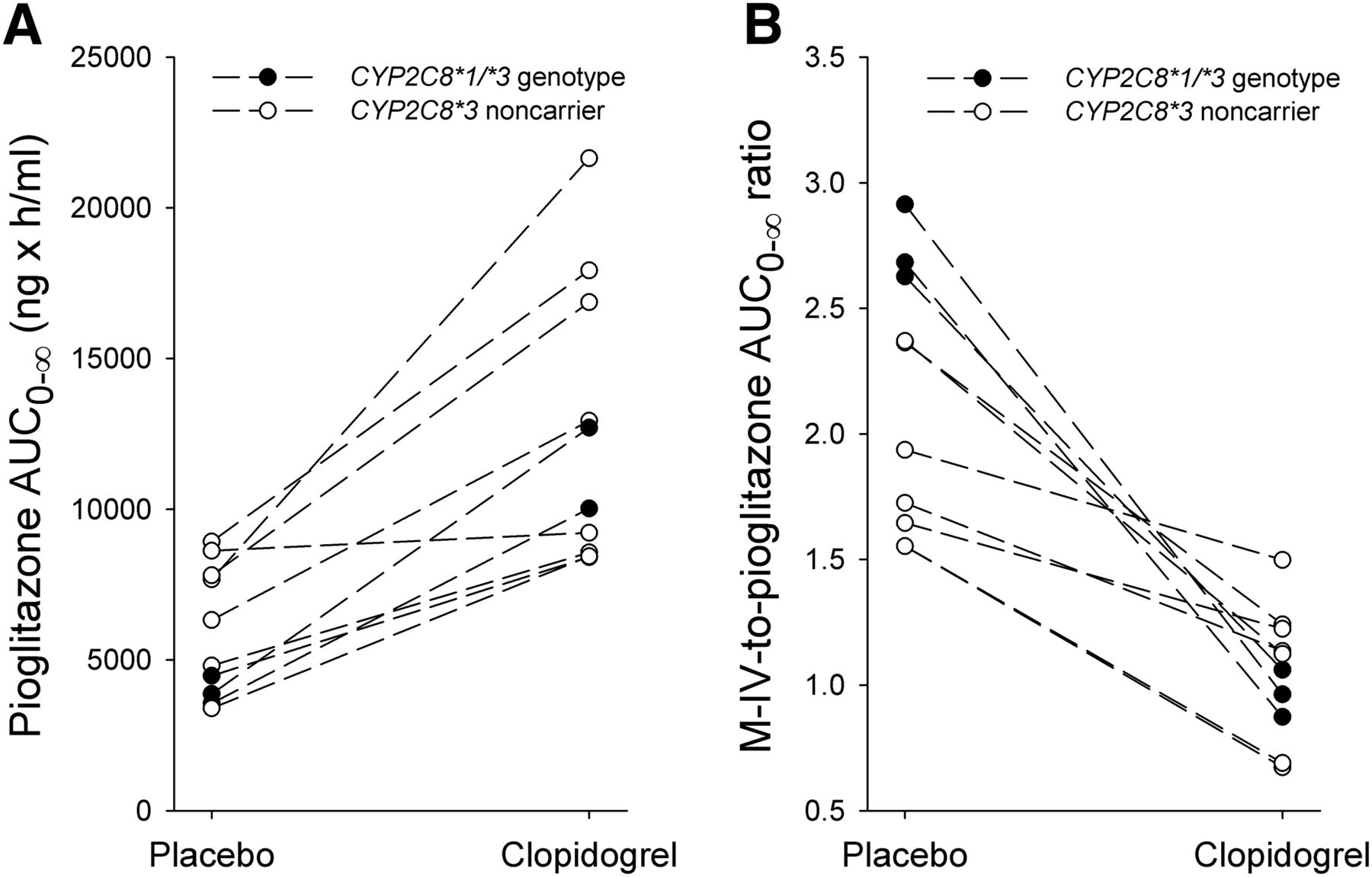
Clopidogrel increased the AUC0–∞ of pioglitazone 2.1-fold (P < 0.001, 90% CI of the geometric mean ratio 1.8–2.6) and prolonged the mean elimination t1/2 of pioglitazone from 6.7 to 11 hours (P = 0.002, 1.7-fold, 90% CI 1.4–2.0), compared with placebo (Fig. 1A; Table 1). The AUC0–∞, and t1/2 of pioglitazone were increased in all 10 subjects by clopidogrel, the largest individual AUC0–∞ increase being 3.3-fold (Fig. 2A). Clopidogrel had no effect on pioglitazone Cmax, but the pioglitazone concentration at 24 hours (C24) was 4.5-fold higher (range 1.6–9.8; P < 0.001) during clopidogrel than during the placebo phase. The tmax occurred later in the clopidogrel phase (P = 0.025).

The effect of clopidogrel (300 mg on day 1, followed by 75 mg on days 2 and 3) on the mean plasma concentrations of pioglitazone (A), its primary metabolite, M-IV (B), and its secondary metabolite, M-III (C). On day 1 of clopidogrel (solid circles) and placebo (open circles) phases, pioglitazone (15 mg) was administered to 10 healthy individuals 1 hour after first dose of pretreatment. Data are presented as geometric means with 90% confidence intervals. For clarity, some error bars have been omitted. Inset depicts the same pioglitazone data on semilogarithmic scale.
Pharmacokinetic variables of pioglitazone and its metabolites M-IV and M-III in 10 healthy volunteers who ingested placebo on days 1, 2, and 3, or 300 mg clopidogrel on day 1, and 75 mg clopidogrel on days 2 and 3, and a single 15-mg dose of pioglitazone 1 hour after the administration of placebo or clopidogrel on day 1
Data are given as geometric mean with geometric coefficient of variation, tmax as median with range. The geometric mean ratios between the two phases are given with 90% CI.

The effect of clopidogrel on individual area under the plasma concentration time curve from zero to infinity (AUC0–∞) of pioglitazone (A) and individual M-IV-to-pioglitazone AUC0–∞ ratio (B) in 10 healthy subjects after ingestion of 15 mg of pioglitazone in a crossover study during the two study phases. Pioglitazone was ingested 1 hour after placebo on day 1 and 1 hour after clopidogrel 300 mg. Open circles, CYP2C8*3 noncarriers (N = 7); solid circles, individuals with the CYP2C8*1/*3 genotype (N = 3). The M-IV-to-pioglitazone metabolic ratio and pioglitazone AUC0–∞ of CYP2C8*3 carriers were compared with those of CYP2C8*3 noncarriers by independent-samples t tests. The subject with the nearly unchanged pioglitazone AUC0–∞ had an exceptionally slow pioglitazone absorption in the clopidogrel phase.
Pioglitazone Metabolites.
The Cmax of the metabolites M-IV and M-III were 61% (P = 0.007, 90% CI 0.48–0.79) and 58% (P = 0.003, 90% CI 0.46–0.74) of that during the placebo phase, respectively (Table 1). However, as the t1/2 of both metabolites was prolonged by clopidogrel, their AUC0–∞ values were not significantly different between the clopidogrel and placebo phases (Table 1). In the clopidogrel phase, the M-IV-to-pioglitazone AUC0–∞ ratio was 49% (P < 0.001, 90% CI 0.40–0.59) of that during the placebo phase.
CYP2C8 Genotype.
One of the subjects had the CYP2C8*1/*2 genotype, three had the CYP2C8*1/*3 genotype, two had the CYP2C8*1/*4 genotype, and the rest (N = 4) had the CYP2C8*1/*1 genotype. The subjects with the CYP2C8*1/*3 genotype had the highest M-IV-to-pioglitazone AUC0–∞ ratio in the placebo phase (P < 0.001) (Fig. 2B). They also had a greater decrease in the M-IV-to-pioglitazone AUC0–∞ ratio, caused by clopidogrel, than did the noncarriers of CYP2C8*3 (P = 0.015), and there was no statistically significant difference between genotypes in this ratio in the clopidogrel phase (P = 0.676) (Fig. 2B).
Pharmacokinetics of Clopidogrel and In Vitro-In Vivo Correlations.
The AUC0–13h of parent clopidogrel, clopidogrel’s active cis-5-thiol metabolite, clopidogrel carboxylic acid, and clopidogrel acyl-β-d-glucuronide varied greatly between individuals (Fig. 3, Table 2). There were no significant correlations between the AUC0–13h of clopidogrel, or its metabolites, and the fold-increase in pioglitazone AUC0–∞ (data not shown). The prediction model indicated that for substrates with an fmCYP2C8 of 60–80%, lower than 1 μM static concentrations of clopidogrel acyl-β-d-glucuronide could produce a similar 2.1-fold increase in AUC as in the present clopidogrel-pioglitazone study (Fig. 4). Assuming 10% unbound fraction, the median Cmax,u and median Cavg,u (0–13 hours) of clopidogrel acyl-β-d-glucuronide after the 300-mg dose of clopidogrel were 0.85 and 0.26 μM, as indicated by the upper and lower borders of the gray area in Fig. 4.

The mean plasma concentrations of clopidogrel (A), and its cis-5-thiol (B), carboxylic acid (C), and acyl-β-d-glucuronide (D) metabolites after clopidogrel 300 mg in 10 healthy volunteers. Data are presented as geometric means with 90% confidence intervals.
Pharmacokinetic variables of clopidogrel, clopidogrel active cis-5-thiol metabolite, clopidogrel carboxylic acid, and clopidogrel acyl-β-d-glucuronide in 10 healthy volunteers after 300 mg (day 1) of clopidogrel, which was ingested 1 hour before pioglitazone
Data are given as median with range.

Static prediction model for multiples of increase in the AUC of pioglitazone (AUCI/AUC) in relation to the plasma concentrations of unbound clopidogrel acyl-β-d-glucuronide, as described in Materials and Methods. The median maximum concentration and the median average concentration (0–13 hours) of clopidogrel acyl-β-d-glucuronide (assuming 10% unbound fraction) after clopidogrel 300 mg were 0.85 and 0.26 μM, respectively, as indicated by the upper and lower borders of the gray area. The fmCYP2C8 expresses the fraction of pioglitazone dose metabolized by CYP2C8. The horizontal dashed gray line represents the observed median fold increase in pioglitazone AUC in the present study.
Discussion
In the present study, clinically used doses of clopidogrel increased the exposure to pioglitazone more than 2-fold, and the C24, corresponding to the trough pioglitazone concentration during a typical once-daily clinical use, was increased 4.5-fold by clopidogrel. In the clopidogrel phase, the metabolite M-IV-to-pioglitazone AUC0–∞ ratio was 49% of that during the placebo phase, consistent with strong inhibition of CYP2C8 by clopidogrel acyl-β-d-glucuronide. The sum of the AUC0–∞ values of pioglitazone, M-IV, and M-III was larger during the clopidogrel than during the placebo phase. Despite some inaccuracies in the extrapolated AUC fractions, this suggests that clopidogrel markedly increases the net exposure to pioglitazone and its active metabolites. This drug-drug interaction is therefore likely to increase the risk of concentration-dependent adverse effects of pioglitazone.
In in vitro experiments, clopidogrel and its metabolites have been shown to inhibit drug metabolizing enzymes, e.g., CYP2B6, CYP2C8, and CYP3A4, and drug transporters, e.g., organic anion–transporting polypeptide (OATP) 1B1, suggesting that clopidogrel can cause changes in the pharmacokinetics of other drugs (Richter et al., 2004; Turpeinen et al., 2005; Floyd et al., 2012; Tamraz et al., 2013; Tornio et al., 2014). Recently, it was reported that clopidogrel use is associated with markedly elevated risk for cerivastatin-induced rhabdomyolysis (Floyd et al., 2012). Also, clopidogrel and its 2-oxo, carboxylic acid and acyl-β-d-glucuronide metabolites were found to inhibit CYP2C8 in vitro, but the inhibitory effects were not strong enough to explain a clinically relevant interaction by clopidogrel (Floyd et al., 2012). In a subsequent study, clopidogrel was found to greatly increase the exposure to the glucose-lowering drug repaglinide (Tornio et al., 2014). In addition, clopidogrel acyl-β-d-glucuronide was identified as a potent time-dependent inhibitor of CYP2C8, whereas clopidogrel and its 2-oxo and carboxylic acid metabolites showed only relatively weak direct inhibition in vitro (Tornio et al., 2014). The active cis-5-thiol metabolite of clopidogrel is very unstable and not commercially available. Therefore, the contribution of this metabolite to CYP2C8 inhibition could not be estimated accurately (Tornio et al., 2014). Both cerivastatin and repaglinide are substrates for CYP2C8, as well as for CYP3A4 and OATP1B1 (Mück, 1998; Kantola et al., 1999; Backman et al., 2002; Wang et al., 2002; Niemi et al., 2003; Kajosaari et al., 2005; Niemi et al., 2005). According to a recent study, clopidogrel does not cause a significant change in the disposition of simvastatin or its active acid form in humans (Itkonen et al., 2015). As simvastatin is among the most sensitive drugs to changes in CYP3A4 and OATP1B1 activity (Neuvonen et al., 1998; Backman et al., 2000; Ichimaru et al., 2001; Prueksaritanont et al., 2003; Pasanen et al., 2006; Niemi et al., 2011), clopidogrel is unlikely to have a clinically relevant effect on CYP3A4 or OATP1B1 activity (Itkonen et al., 2015). These findings indicate that inhibition of CYP2C8 by clopidogrel acyl-β-d-glucuronide is the main mechanism of the clopidogrel-cerivastatin and clopidogrel-repaglinide interactions (Floyd et al., 2012; Tornio et al., 2014; Itkonen et al., 2015). This conclusion is also supported by a recent case report, which suggests that clopidogrel significantly reduces the metabolic clearance of the CYP2C8 substrate anticancer agent paclitaxel (Bergmann et al., 2016).
According to in vitro studies, pioglitazone can be metabolized by several cytochrome P450 enzymes, e.g., CYP2C8 and CYP3A4 (Jaakkola et al., 2006a; Scheen, 2007; Tornio et al., 2012). However, the strong CYP3A4 inhibitor itraconazole has had no effect on the pharmacokinetics of pioglitazone, whereas the strong CYP2C8 inhibitor gemfibrozil has increased the AUC0–∞ of pioglitazone 3- to 4-fold (Deng et al., 2005; Jaakkola et al., 2005; Aquilante et al., 2013b). Also trimethoprim, which is a weak inhibitor of CYP2C8, has raised the exposure to pioglitazone 1.4-fold (Tornio et al., 2008). Thus, in vivo in humans, CYP2C8 is a crucial enzyme in the biotransformation of pioglitazone, accounting for about 70–75% of the total elimination of pioglitazone, and the contribution of CYP3A4 to its elimination seems to be negligible. Furthermore, the pharmacokinetics of pioglitazone are not affected by the SLCO1B1 c.521T>C polymorphism, indicating that pioglitazone is not a substrate of OATP1B1 (Kalliokoski et al., 2008). Therefore, pioglitazone can be used as a selective probe substrate to study the function of CYP2C8 in vivo (Tornio et al., 2012; Backman et al., 2016). The average 2.1-fold increase in pioglitazone AUC by clopidogrel in the current study is in agreement with the results of earlier drug interaction studies between pioglitazone and CYP2C8 inhibitors (Deng et al., 2005; Jaakkola et al., 2005; Tornio et al., 2008). The result also conforms well with the recent prediction that daily dosing of clopidogrel 75 mg in steady state causes 60–85% inhibition of CYP2C8 activity (Tornio et al., 2014).
The previous static predictions of the effects of clopidogrel and its metabolites on CYP2C8 activity indicated that during standard dosing of clopidogrel, the reversible CYP2C8 inhibitory effects of clopidogrel and clopidogrel carboxylic acid are negligible compared with the mechanism-based CYP2C8-inhibitory effect of clopidogrel acyl-β-d-glucuronide (Tornio et al., 2014). Accordingly, we used the reported mechanism-based CYP2C8 inhibitory constants of the glucuronide metabolite to predict the effect of clopidogrel on the AUC of a CYP2C8 substrate with an fmCYP2C8 of 60–80%. On the basis of studies with gemfibrozil (Deng et al., 2005; Jaakkola et al., 2005; Aquilante et al., 2013b), this fmCYP2C8 range probably includes the fmCYP2C8 of pioglitazone in most individuals. Our predictions showed that clopidogrel acyl-β-d-glucuronide concentrations corresponding to its peak or average (0–13 hours) unbound plasma concentrations after a 300-mg clopidogrel dose can cause an AUC increase similar to that observed in the present study. It should be noted, however, that this was not a static steady-state study, and that owing to the 75-mg doses of clopidogrel given on days 2 and 3, the concentrations of clopidogrel acyl-β-d-glucuronide during most of the 72-hour study period were much lower than the average 0- to 13-hour concentration. Therefore, it is likely that the unbound concentrations of clopidogrel acyl-β-d-glucuronide are much higher at the CYP2C8 enzyme site than in plasma.
Although clopidogrel increased the AUC0–∞ of pioglitazone, it did not increase pioglitazone Cmax. This could be explained by the high oral bioavailability and limited first-pass metabolism of pioglitazone (Eckland and Danhof, 2000). Similarly, a 3-day pretreatment with the strong time-dependent CYP2C8 inhibitor gemfibrozil had no effect on pioglitazone Cmax, even though it increased the AUC0–∞ by 3.2-fold (Jaakkola et al., 2005). It should be noted that CYP2C8 inhibition by clopidogrel probably takes place quite rapidly, despite the time-dependent mechanism (Tornio et al., 2014). Also the inactivation of CYP2C8 by gemfibrozil (via formation of an acyl glucuronide metabolite) is a rapid process, as indicated by a 90% decrease in the AUC of a CYP2C8-dependent metabolite, and a nearly maximum increase in the AUC of the CYP2C8 substrate repaglinide when ingested 1 hour after 600 mg of gemfibrozil (Honkalammi et al., 2011). Theoretically, a prolonged absorption or an increased distribution volume of pioglitazone, together with its impaired metabolism, could also explain the unchanged Cmax despite the increased AUC0–∞.
The Cmax values of pioglitazone metabolites IV and III occurred considerably later than the Cmax of pioglitazone. Consistent with impaired and delayed metabolite formation, clopidogrel prolonged further the tmax values of these metabolites. Yet, because of the prolonged half-lives of these metabolites, their AUC0–∞ values were not changed by clopidogrel. Interestingly, in the study of Jaakkola et al. (2005), gemfibrozil caused changes very similar to those caused by clopidogrel in the time-courses of pioglitazone and its M-IV and M-III metabolite concentrations. Thus, it is reasonable to assume also that the further metabolism of M-IV and M-III is partially dependent on the activity of CYP2C8.
The increase in the AUC0–∞ of pioglitazone by clopidogrel varied between 1.1- and 3.3-fold in the homogenous group of 10 healthy volunteers. However, the extent of the interaction did not correlate significantly with the plasma concentrations (AUC0–13h) of clopidogrel, or its cis-5-thiol, carboxylic acid, and acyl-β-d-glucuronide metabolites. Previous studies have shown that the AUC of pioglitazone is about 30% lower in CYP2C8*3 carriers than in noncarriers, and that gemfibrozil increases pioglitazone exposure somewhat more in CYP2C8*3 carriers than in noncarriers (Tornio et al., 2008; Aquilante et al., 2013a, b; Kadam et al., 2013). In the present study, the AUC of pioglitazone was about 40% lower in the three individuals with the CYP2C8*1/*3 genotype than in CYP2C8*3 noncarriers during the placebo phase. Furthermore, the M-IV-to-pioglitazone AUC0–∞ ratio, an index of the CYP2C8-mediated metabolic clearance of pioglitazone, was highest in these three individuals. The inhibitory effect of clopidogrel on this metabolic ratio was greatest in carriers of the CYP2C8*3 allele, and consequently the difference between genotypes in the metabolic ratio disappeared in the clopidogrel phase. This finding is biologically plausible, since the contribution of CYP2C8 to the metabolism of pioglitazone should be greatest in individuals carrying the increased activity CYP2C8*3 allele. Overall, these findings suggest that carriers of the CYP2C8*3 allele are more susceptible than noncarriers to the interaction between clopidogrel and pioglitazone. However, it should be noted that the sample size of the present study was not sufficient to draw any firm conclusions concerning these genotype effects, and a clinical study with an adequate statistical power is needed to confirm this hypothesis.
The results of the current study have clear clinical implications, although the study was performed in healthy volunteers. The 300-mg loading dose of clopidogrel followed by 75 mg daily is the recommended dosing regimen. Only a single low 15-mg therapeutic dose of pioglitazone was administered for safety reasons. However, the pharmacokinetics of pioglitazone is linear and similar in patients with type 2 diabetes mellitus to that in healthy volunteers (Eckland and Danhof, 2000). Moreover, it is unlikely that there would be a major difference in clopidogrel pharmacokinetics between healthy subjects and patients with diabetes. Thus, the result of our pharmacokinetic interaction study, i.e., markedly increased pioglitazone exposure by clopidogrel, should be transferable to diabetic patients irrespective of the pioglitazone dose.
Pioglitazone has a generally favorable side-effect profile, but like rosiglitazone, it can cause dose-related fluid retention, and this is more likely to occur when pioglitazone is used in combination with insulin or sulphonylureas (Nesto et al., 2003). Fluid retention, in turn, can worsen the symptoms of congestive heart failure and even cause pulmonary edema (Waugh et al., 2006). Pioglitazone, either alone or in combination with other glucose-lowering drugs, is often used in conjunction with ADP-receptor inhibitors, e.g., clopidogrel, in the treatment of patients with type 2 diabetes mellitus with cardiovascular comorbidity. It is reasonable to assume that addition of clopidogrel to a pioglitazone-containing drug regimen can markedly raise pioglitazone exposure, in some patients by even more than 3-fold. Furthermore, as clopidogrel prolongs the t1/2 of pioglitazone, its trough concentrations would probably increase even more, up to 10-fold. At least theoretically, the uninterrupted high pioglitazone exposure could increase the risk of fluid retention even more than the daily fluctuating exposure, which includes low concentrations before the next dosing.
Glucuronide metabolites have only recently gained attention as perpetrators of drug-drug interactions (Backman et al., 2016). In addition to clopidogrel acyl-β-d-glucuronide, gemfibrozil 1-O-β-glucuronide and deleobuvir acyl glucuronide (M829/2) have been identified as time-dependent inhibitors of CYP2C8 (Shitara et al., 2004; Ogilvie et al., 2006; Tornio et al., 2014; Sane et al., 2016). The active site of CYP2C8 is relatively large (Schoch et al., 2004) and can accommodate these glucuronide metabolites, allowing their metabolism to a reactive species that inactivates CYP2C8. The present findings corroborate the previous report of the CYP2C8-mediated clopidogrel-repaglinide interaction, in the sense that clopidogrel, via its glucuronide metabolite, is able to cause strong inhibition of CYP2C8 activity. Thus, glucuronide metabolites of some other drugs may also cause unexpected drug interactions with pioglitazone and other CYP2C8 substrates (Backman et al., 2016).
In conclusion, our findings indicate that clopidogrel is a clinically relevant CYP2C8 inhibitor, and concomitant use of clopidogrel and pioglitazone may therefore increase the efficacy and risk of concentration-related adverse effects of pioglitazone. Thus, the combination of pioglitazone and clopidogrel should be used with caution, and dose reduction of pioglitazone should be considered if these two drugs are used concomitantly. Clopidogrel is likely to increase the exposure to other drugs mainly metabolized by CYP2C8, e.g., rosiglitazone.
Acknowledgments
The authors thank Jouko Laitila, Eija Mäkinen-Pulli, and Lisbet Partanen for their technical assistance.
Authorship Contributions
Participated in research design: Itkonen, Tornio, Neuvonen, Neuvonen, Niemi, Backman.
Conducted experiments: Itkonen, Tornio, Neuvonen, Neuvonen, Niemi, Backman.
Contributed new reagents or analytic tools: Itkonen, Tornio, Neuvonen, Neuvonen, Niemi, Backman.
Performed data analysis: Itkonen, Tornio, Neuvonen, Neuvonen, Niemi, Backman.
Wrote or contributed to the writing of the manuscript: Itkonen, Tornio, Neuvonen, Neuvonen, Niemi, Backman.
Footnotes
- Received March 7, 2016.
- Accepted June 2, 2016.
This work was supported by grants from the Academy of Finland [Grant decision 278123, 2014], and the Helsinki University Central Hospital Research Fund (Helsinki, Finland).
Abbreviations
- AUC
- area under the plasma concentration-time curve
- CI
- confidence interval
- Cmax
- peak plasma concentration
- fmCYP2C8
- fraction metabolized by CYP2C8
- M-III
- ketopioglitazone
- M-IV
- hydroxypioglitazone
- OATP
- organic anion–transporting polypeptide
- tmax
- time to Cmax
- t1/2
- half-life
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}