


Abstract
Growing evidence suggests that certain glucuronides function as potent inhibitors of CYP2C8. We previously reported the possibility of drug-drug interactions between candesartan cilexetil and paclitaxel. In this study, we evaluated the effects of candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide on pathways associated with the elimination of paclitaxel, including those involving organic anion–transporting polypeptide (OATP) 1B1, OATP1B3, CYP2C8, and CYP3A4. UDP-glucuronosyltransferase (UGT) 1A10 and UGT2B7 were found to increase candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide formation in a candesartan concentration–dependent manner. Additionally, the uptake of candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide by cells stably expressing OATPs is a saturable process with Km of 5.11 and 12.1 μM for OATP1B1 and 28.8 and 15.7 μM for OATP1B3, respectively; both glucuronides exhibit moderate inhibition of OATP1B1/1B3. Moreover, the hydroxylation of paclitaxel was evaluated using recombinant CYP3A4 and CYP3A5. Results show that candesartan, candesartan N2-glucuronide, and candesartan acyl-β-D-glucuronide inhibit the CYP2C8-mediated metabolism of paclitaxel, with candesartan acyl-β-D-glucuronide exhibiting the strongest inhibition (IC50 is 18.9 µM for candesartan acyl-β-D-glucuronide, 150 µM for candesartan, and 166 µM for candesartan N2-glucuronide). However, time-dependent inhibition of CYP2C8 by candesartan acyl-β-D-glucuronide was not observed. Conversely, the IC50 values of all the compounds are comparable for CYP3A4. Taken together, these data suggest that candesartan acyl-β-D-glucuronide is actively transported by OATPs into hepatocytes, and drug-drug interactions may occur with coadministration of candesartan and CYP2C8 substrates, including paclitaxel, as a result of the inhibition of CYP2C8 function.
Significance Statement This study demonstrates that the acyl glucuronidation of candesartan to form candesartan acyl-β-D-glucuronide enhances CYP2C8 inhibition while exerting minimal effects on CYP3A4, organic anion–transporting polypeptide (OATP) 1B1, and OATP1B3. Thus, candesartan acyl-β-D-glucuronide might represent a potential mediator of drug-drug interactions between candesartan and CYP2C8 substrates, such as paclitaxel, in clinical settings. This work adds to the growing knowledge regarding the inhibitory effects of glucuronides on CYP2C8.
Introduction
Glucuronidation of drugs is involved in their detoxification in mammals. Acyl glucuronides have been reported as inducers of toxicity owing to their high chemical reactivity, which, for example, causes acylation and glycation of proteins, resulting in their covalent binding to proteins (Lassila et al., 2015). Some acyl glucuronides can interact with CYP isoforms. In particular, growing evidence suggests that some glucuronides are potent inhibitors of CYP2C8, an important enzyme involved in drug metabolism (Backman et al., 2016; Tornio et al., 2017). In fact, the role of glucuronides in drug-drug interactions via CYP2C8 has been recognized for combination therapy with cerivastatin and gemfibrozil (Chang et al.., 2004). However, severe myopathy and rhabdomyolysis result from drastically altered pharmacokinetics of cerivastatin (Backman et al., 2002) due to potent inhibition of CYP2C8 by gemfibrozil acyl-β-D-glucuronide (Backman et al., 2002; Wang et al., 2002; Ogilvie et al., 2006; Takagi et al., 2015; Varma et al., 2015). Similarly, coadministration of clopidogrel with repaglinide induces an approximately 2.5–5.1-fold increase in the area under the plasma concentration-time curve of repaglinide (Tornio et al., 2014), which is a substrate of CYP2C8 and organic anion–transporting polypeptide (OATP) 1B1, a hepatic uptake transporter (Kajosaari et al., 2005; Gertz et al., 2014). Orally administered clopidogrel is hydrolyzed to clopidogrel carboxylic acid and subsequently converted to clopidogrel acyl-β-D-glucuronide (Farid et al., 2010), which is a strong, time-dependent inhibitor of CYP2C8 and a weak inhibitor of OATP1B1 that is considered to be responsible for the interaction between repaglinide and clopidogrel (Tornio et al., 2014). Besides gemfibrozil and clopidogrel, numerous other drug substrates have been described for glucuronosyltransferases; however, the CYP2C8 inhibitory effects of other glucuronides remain unknown.
After intravenous administration, paclitaxel is transported to hepatocytes by OATP1B1 and OATP1B3, where it becomes metabolized to 6α-hydroxypaclitaxel and p-3′-hydroxypaclitaxel by CYP2C8 and to a lesser extent by CYP3A4, respectively (Fujino et al., 2001; Cresteil et al., 2002). Agergaard et al. (2017) reported that concomitant use of clopidogrel aggravated paclitaxel-induced neuropathy in patients, thereby confirming that coadministration of paclitaxel with medications exerting CYP2C8 inhibitory effects results in severe adverse events caused by drug-drug interactions.
Previously, we performed a comprehensive retrospective analysis of concomitant medications to identify potential risk factors for chemotherapy-induced severe neutropenia in patients administered paclitaxel plus carboplatin combination therapy. We found that concomitant use of candesartan cilexetil, unlike that of other angiotensin receptor blockers, could result in severe neutropenia (Katsube et al., 2018). Candesartan cilexetil, an angiotensin II receptor blocker, is commonly administered for the treatment of hypertension. Almost all candesartan cilexetil is hydrolyzed to candesartan in the gastrointestinal tract (Yamaoka et al., 1981). However, previous in vitro studies have shown that the inhibitory effects of candesartan on CYP2C8/CYP3A4 and OATPs are not sufficiently strong to suggest an interaction with paclitaxel at clinically relevant concentrations (Taavitsainen et al., 2000; Senda et al., 2015; van de Steeg et al., 2015). After oral administration of candesartan to rats and dogs, candesartan acyl-β-D-glucuronide and candesartan N2-glucuronide were detected in the feces and blood (Kondo et al., 1996), respectively (Fig. 1). In addition, glucuronidation of candesartan by human UDP-glucuronosyltransferase (UGT) yields two metabolites identified as O-glucuronide and tetrazole-N2-glucuronide when candesartan is incubated with human liver microsomes (HLMs) or with individual human UGTs (Alonen et al., 2008). However, no definitive in vitro proof of interactions between candesartan glucuronides and the paclitaxel hepatic elimination pathways, including those involving OATPs and CYPs, has been presented.

Chemical structures of candesartan cilexetil and its metabolites.
The objective of the present study was, therefore, to investigate the possible interactions of candesartan and candesartan glucuronides with CYP2C8. We also sought to determine whether candesartan glucuronides serve as substrates of OATPs and as inhibitors of CYP2C8, CYP3A4, and OATPs in vitro.
Materials and Methods
Materials.
Paclitaxel and glucose-6-phosphate dehydrogenase (G6PDH) were purchased from Wako Pure Chemical Industries (Osaka, Japan). β-NADP+ and glucose-6-phosphate (G6P) were purchased from Oriental Yeast Co. (Tokyo, Japan). Docetaxel trihydrate, 2′,7′-dichlorofluorescein (DCF), and fluorescein (FL) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Candesartan was purchased from LKT Laboratories, Inc. (St. Paul, MN). Candesartan N2-glucuronide, candesartan acyl-β-D-glucuronide, and 6α-hydroxypaclitaxel were purchased from Toronto Research Chemicals (North York, ON, Canada). HLMs were purchased from BD Biosciences (Woburn, MA). These HLMs were pooled from 21 individuals (14 males and 7 females). Human recombinant CYP2C8 and CYP3A4 were purchased from Cypex (Dundee, UK). p-3′-Hydroxypaclitaxel and human recombinant UGT1A10 and UGT2B7 were purchased from Corning (Corning, NY). UDP–glucuronic acid (UDPGA) was purchased from Yamasa Co. (Tokyo, Japan).
Cells.
Previously established OATP1B1-transfected human embryonic kidney (HEK) 293 (HEK/OATP1B1) cells and empty vector–transfected HEK293 (HEK/Mock) cells (Katsube et al., 2017) were used in this study. OATP1B3-transfected HEK293 (HEK/OATP1B3) cells were kindly provided by Professor Hiroyuki Kusuhara and Dr. Kazuya Maeda (Laboratory of Molecular Pharmacokinetics, Tokyo University, Japan). HEK/OATP1B1, HEK/OATP1B3, and HEK/Mock cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Grand Island, NY) and supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific, Waltham, MA), 100 U/ml penicillin, 100 µg/ml streptomycin (Nakalai Tesque, Kyoto, Japan), 0.1 mM nonessential amino acids (Nakalai Tesque), and 0.72 mM geneticin (Nakalai Tesque) in an atmosphere of 5% CO2 in a humidified incubator at 37°C.
For cellular uptake experiments, cells were seeded in collagen-coated 48-well (0.4 × 105 cells/well) or 24-well (0.8 × 105 cells/well) plates (Corning). The cells were cultured in DMEM in a humidified atmosphere with 5% CO2 at 37°C. After 3 days, the medium was replaced with DMEM containing 5 mM sodium butyrate, a histone deacetylase inhibitor, to enhance gene expression (Kondo et al., 1996). On the next day, the intracellular uptake experiments described below were performed.
Inhibition of OATP1B1 and OATP1B3 Activity.
The intracellular uptake experiments were performed as previously described (Katsube et al., 2017) with DCF and FL used as OATP1B1 and OATP1B3 substrates, respectively (De Bruyn et al., 2011; Koide et al., 2018). The cells were washed twice with warmed HEPES/Hank’s balanced salt solution (HBSS) (137 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 0.4 mM MgSO4, 0.5 mM MgCl2, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 4.2 mM NaHCO3, 5.6 mM glucose, and 25.0 mM HEPES, pH 7.4) and then preincubated for 10 minutes at 37°C in HEPES/HBSS buffer. After aspiration of the HEPES/HBSS buffer, the uptake assay was initiated by the addition of 0.2 ml of warmed HEPES/HBSS buffer containing the substrate (1 µM DCF or 3 µM FL) with or without the test compound (candesartan, candesartan N2-glucuronide, or candesartan acyl-β-D-glucuronide) at concentrations of 1 and 10 µM. The uptake assay was terminated by removing the uptake buffer 3 minutes after initiation, and the cells were subsequently washed three times with ice-cold PBS containing 0.5 mM MgCl2 and 1 mM CaCl2.
To quantify the intracellular accumulation of DCF and FL, the cells were lysed with 150 µl of 0.1 M NaOH. To normalize the data to cell numbers, we measured the protein content using the Lowry protein assay with bovine serum albumin as the standard (Lowry et al., 1951). FL and DCF were quantified as described previously (Koide et al., 2018). Briefly, 100 μl of each sample was transferred to a 96-well solid black polystyrene plate (Corning), and quantification was carried out by measuring the fluorescence intensity (excitation at 490 nm; emission at 515 nm) using a microplate reader (Power Scan HT; BioTek Japan, Tokyo, Japan).
Intracellular Glucuronide Uptake Mediated by OATP1B1 or OATP1B3.
In the intracellular uptake experiments, HEK/OATP1B1 or HEK/OATP1B3 cells were washed twice with warmed HEPES/HBSS buffer and then preincubated for 10 minutes at 37°C in HEPES/HBSS buffer. After aspiration of the HEPES/HBSS buffer, uptake was initiated via addition of warmed HEPES/HBSS buffer containing candesartan N2-glucuronide or candesartan acyl-β-D-glucuronide (0.5–30 µM). The uptake experiments were terminated by removal of the uptake buffer 3 minutes after initiation; the cells were then washed three times with ice-cold PBS containing 0.5 mM MgCl2 and 1 mM CaCl2.
To quantify the intracellular accumulation of glucuronides, cells were lysed with PBS containing 0.1% (w/v) Tween 20. The data were normalized to cell number as described above, and the glucuronides were quantified as described previously (Alonen et al., 2008). Briefly, 150 µl of acetonitrile containing 267 nM olmesartan as an internal standard (Ma et al., 2005) and 230 µl of 50 mM phosphate buffer (pH 3.0) were added to 110 µl of the sample. The mixture was vigorously vortexed for 1 minute and centrifuged at 15,000g for 10 minutes, after which the upper layer was injected into an Inertsil ODS-III column (5 µm, 250 × 4.0 mm i.d.; GL Sciences, Inc., Tokyo, Japan), and the glucuronides were quantified using a high-performance liquid chromatography (HPLC) system equipped with a fluorescence detector (RF-20A; Shimadzu, Kyoto, Japan) by measuring fluorescence intensity (excitation at 260 nm; emission at 395 nm). For HPLC, the flow rate of the mobile phase comprising 50 mM phosphate buffer (pH 3.0) and acetonitrile (70:30, v/v) was 1.0 ml/min.
Glucuronidation of Candesartan by Recombinant Human UGT1A10 and UGT2B7.
Relatively high levels of O-glucuronidation corrected for the expression of individual human recombinant UGTs were obtained, with the highest levels observed with UGT1A10 followed by UGT1A7–1A9. In addition, relatively high levels of N2-glucuronidation were obtained using UGT1A3 and UGT2B7 when candesartan and recombinant protein were incubated for 4 hours at concentrations of 1 mM and 1.0 mg protein/ml, respectively (Alonen et al., 2008). Thus, candesartan glucuronidation was assessed using human recombinant UGT1A10 and UGT2B7 as described previously (Alonen et al., 2008), with slight modifications. Each reaction mixture contained candesartan (0.5–30 µM) and 0.5 mg·ml−1 human recombinant UGT1A10 or UGT2B7, 27.5 µg·ml−1 alamethicin, and 50 mM phosphate buffer with 10 mM Mg2+ (pH 7.4). The reaction mixture was incubated at 37°C for 5 minutes, and the reaction was initiated by the addition of 5 mM UDPGA. The mixture was incubated at 37°C for another 60 minutes; thereafter, the reaction was terminated by the addition of acetonitrile. Glucuronides were quantified according to the method described above.
Inhibition of Paclitaxel Metabolism in Human Liver Microsomes or Recombinant Human CYP2C8 and CYP3A4.
Hydroxylation of paclitaxel was assessed using HLMs or human recombinant CYP2C8 and CYP3A4 as previously described (Wang et al., 2014; Tsujimoto et al., 2016), with minor modifications. Each reaction mixture contained 10 pmol/ml recombinant CYP2C8 or CYP3A4 with or without the test compounds (15.6–250 µM) in 50 mM phosphate buffer with 5 mM Mg2+ (pH 7.4). The CYP inhibition experiments were conducted through the following methods. First, with reversible inhibition, to determine the IC50 values of the test compounds by direct CYP inhibition, the reaction mixture was incubated at 37°C for 5 minutes, after which the reaction was initiated by addition of the NADPH-regenerating system (5 mM G6P, 1.0 U/ml G6PDH, and 1 mM β-NADP+) containing paclitaxel (2 µM). The mixture was then incubated at 37°C for 30 minutes and subsequently terminated by adding acetonitrile. Second, with IC50 shift assay, the time-dependent inhibition (TDI) of CYP was evaluated using the IC50 shift assay, which is the most commonly used assay for the assessment of TDI owing to its relative ease of execution in a drug discovery setting (Orr et al., 2012). To perform the IC50 shift assay, the reaction mixture (0.1 mg/ml HLMs, the NADPH-regenerating system, and a test compound in 50 mM phosphate buffer with 5 mM Mg2+, pH 7.4) was incubated at 37°C with the initial addition of the NADPH-regenerating system for 30 minutes. The mixture was then added to a paclitaxel (2 µM)-containing solution and incubated for 10 minutes; the reaction was terminated by the addition of acetonitrile. Third, with Ki experiment, the Ki values for CYP2C8 were determined using the Dixon plot (no initial inhibition). Briefly, for direct CYP inhibition by the test compounds, the reaction mixture was incubated at 37°C for 5 minutes, and this was followed by initiation of the reaction by addition of the NADPH-regenerating system (5 mM G6P, 1.0 U/ml G6PDH, and 1 mM β-NADP+) containing paclitaxel (0.5, 1.0, and 2.0 µM). The mixture was then incubated at 37°C for 30 minutes, and the reaction was terminated by adding acetonitrile.
6α-Hydroxypaclitaxel and 3′-p-hydroxypaclitaxel were quantified as described previously (Harris et al., 1994; Huizing et al., 1995). Briefly, 50 µl of acetonitrile containing 1 µM docetaxel as an internal standard (Garg and Ackland, 2000) and 3 ml of diethyl ether were added to a mixture containing the samples and vigorously vortexed for 1 minute. After centrifugation at 1650g for 10 minutes, 2.8 ml of the upper layer was collected and evaporated to dryness under a stream of N2 at 40°C. The residue was then dissolved in the mobile phase (50 mM phosphate buffer, pH 7.0: acetonitrile, 55:45, v/v). The sample was injected into an HPLC system and quantified by measuring the absorbance at 229 nm using an Inertsil ODS-III column (5 µm, 250 × 4.0 mm i.d.) and a fluorescent detector (SPD-20A; Shimadzu).
Data Analysis.
The OATP1B1- or OATP1B3-mediated uptake of the glucuronides was calculated by subtracting the level of intracellular accumulation of the substrates in HEK/Mock cells from that in HEK/OATP1B1 or HEK/OATP1B3 cells. The data are presented as mean ± S.D. of the sample or as estimated mean [95% confidence interval (CI)]. Significant differences between mean values were determined using the Student’s t test or one-way ANOVA followed by the Dunnett’s test, and P < 0.05 was considered statistically significant.
The Michaelis-Menten constant (Km) and Vmax values used as kinetic parameters of the OATP activity were calculated using equations of the Michaelis-Menten model (eq. 2) to analyze the data with the MULTI program (Yamaoka et al., 1981). Equation 1 was used for HEK/OATP1B1 and HEK/OATP1B3 cells: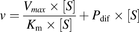 (1)and eq. 2 was used for HEK/Mock cells:
(1)and eq. 2 was used for HEK/Mock cells: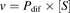 (2)The substrate concentration at which the reaction rate was half of Vmax (S50) and Vmax values used as kinetic parameters of the UGT activity was calculated using the Michaelis-Menten or the Hill eq. 3 to analyze the data with the MULTI program (Yamaoka et al., 1981). The appropriate model was determined using the Eadie-Hofstee plot. When a bell-shaped curve was observed, the data were fitted using the Hill equation, as this is characteristic of sigmoidal data (Madan et al., 2002).
(2)The substrate concentration at which the reaction rate was half of Vmax (S50) and Vmax values used as kinetic parameters of the UGT activity was calculated using the Michaelis-Menten or the Hill eq. 3 to analyze the data with the MULTI program (Yamaoka et al., 1981). The appropriate model was determined using the Eadie-Hofstee plot. When a bell-shaped curve was observed, the data were fitted using the Hill equation, as this is characteristic of sigmoidal data (Madan et al., 2002).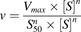 (3)wherein v, [S], and n represent the initial glucuronidation velocity, the substrate concentration, and the Hill coefficient, respectively.
(3)wherein v, [S], and n represent the initial glucuronidation velocity, the substrate concentration, and the Hill coefficient, respectively.
IC50 values of the test compound for the CYP2C8 or CYP3A4 activity were calculated using eq. 4 to analyze the data with the MULTI program (Yamaoka et al., 1981): (4)wherein I, [I], and γ represent the inhibition of CYP activity (% of control), the concentration of the test compound, and a sigmoidal constant, respectively.
(4)wherein I, [I], and γ represent the inhibition of CYP activity (% of control), the concentration of the test compound, and a sigmoidal constant, respectively.
Results
Glucuronidation of Candesartan to N2-Glucuronide and Acyl-β-D-Glucuronide by Recombinant UGT1A10 and UGT2B7.
The concentration of candesartan N2-glucuronide was linearly increased by UGT1A10 and UGT2B7 in a candesartan concentration–dependent manner up to 1 mM (Fig. 2). The kinetics of candesartan glucuronidation to candesartan acyl-β-D-glucuronide by UGT1A10 followed the sigmoidal autoactivation model best fit with the Hill equation (Fig. 2C), which is demonstrated by a bell-shaped curve (Fig. 2C insets), whereas glucuronidation of candesartan to candesartan acyl-β-D-glucuronide by UGT2B7 followed the Michaelis-Menten model.

Kinetic analysis of candesartan glucuronidation to the N2-glucuronide (A and B) and acyl-β-D-glucuronide (C and D) form by human recombinant UGT1A10 and UGT2B7. (A and B) Candesartan N2-glucuronide levels were determined after incubation of the recombinant proteins (1.0 mg/ml) with UDPGA (5 mM) and candesartan (1–5 mM) at 37°C for 60 minutes after a 5-minute preincubation. (C and D) Candesartan acyl-β-D-glucuronide levels were determined after incubation of the recombinant proteins (0.5 mg/ml) with UDPGA (5 mM) and candesartan (30–1000 µM) at 37°C for 60 minutes after a 5-minute preincubation. Each point represents the mean ± S.D. (n = 3).
Uptake of Glucuronides by HEK/OATP1B1 and HEK/OATP1B3 Cells.
The intracellular uptake of candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide by HEK/OATP1B1 and HEK/OATP1B3 cells increased in a concentration-dependent manner and was significantly higher than the uptake by HEK/Mock cells. The OATP1B1- and OATP1B3-mediated uptake of candesartan glucuronide followed the Michaelis-Menten profile. Candesartan N2-glucuronide was transported by OATP1B1 (Km = 5.11 µM) and, to a smaller extent, by OATP1B3 (Km = 28.8 µM). The affinity of acyl glucuronide for OATP1B1 and OATP1B3 showed similar Km values (Km = 12.1 and 15.7 µM, respectively). The Vmax values of OATP1B1 for acyl glucuronide and N2-glucuronide were 0.5 and 1.64 pmol·min−1·mg protein−1, respectively. The Vmax values of OATP1B3 for acyl glucuronide and N2-glucuronide were 3.76 and 3.95 pmol·min−1·mg protein−1, respectively (Fig. 3).

Kinetic analysis of candesartan N2-glucuronide (A) and acyl-β-D-glucuronide (B) uptake mediated by OATP1B1 and OATP1B3. The levels of candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide were determined after incubation of candesartan (0.5–30 µM) with HEK/OATP1B1 and HEK/OATP1B3 cells at 37°C for 1 minute after a 5-minute preincubation. The Km and Vmax values were determined from a Michaelis-Menten plot using the MULTI program. Each point represents the mean ± S.D. (n = 4). The Km and Vmax values represent the estimate with 95% CI.
Inhibitory Effects of Candesartan and Its Glucuronides on the Uptake of OATP1B1 and OATP1B3 Substrates.
Candesartan and its glucuronides significantly inhibited the OATP1B1-mediated uptake of DCF and the OATP1B3-mediated uptake of FL in a concentration-dependent manner. OATP1B1- or OATP1B3-mediated uptake of each substrate was reduced by approximately 30%–40% in the presence of 10 µM candesartan and candesartan acyl glucuronide compared with the control. In the presence of 10 µM candesartan N2-glucuronide, OATP1B1-mediated uptake of DCF was reduced by 66.7% compared with that in the control, whereas OATP1B3-mediated uptake of FL was only reduced by 36.3 (Fig. 4).

Effects of candesartan and its glucuronides on the uptake of OATP1B1 and OATP1B3 probe substrates. (A) OATP1B1-mediated uptake of DCF was determined after incubation of DCF (5 µM) in the absence (control) or presence of candesartan (1 or 10 µM), candesartan N2-glucuronide (1 or 10 µM), or acyl-β-D-glucuronide (1 or 10 µM) with HEK/OATP1B1 cells. (B) OATP1B3-mediated uptake of FL was determined after incubation of FL (2 µM) in the absence (control) or presence of candesartan (1 or 10 µM), candesartan N2-glucuronide (1 or 10 µM), or acyl-β-D-glucuronide (1 or 10 µM) with HEK/OATP1B3 cells. Each point represents the mean ± S.D. (n = 4). The significance of any differences from the control was determined by ANOVA followed by the Dunnett’s test (*P < 0.01, **P < 0.001).
Inhibitory Effects of Candesartan and Its Glucuronides on the Hydroxylation of Paclitaxel by Recombinant CYP2C8 and CYP3A4.
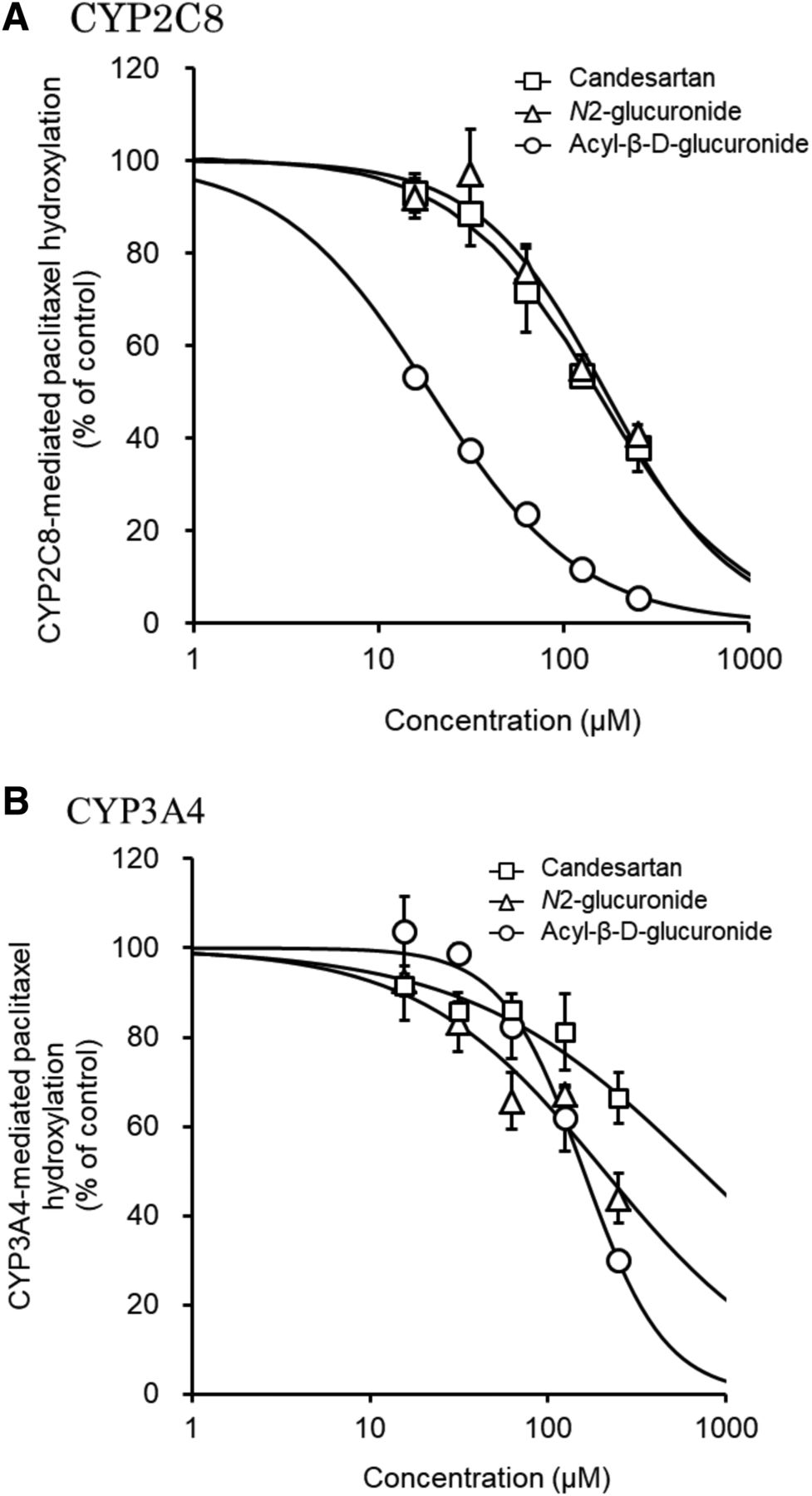
Candesartan, candesartan N2-glucuronide, and candesartan acyl-β-D-glucuronide inhibited the CYP2C8-mediated metabolism of paclitaxel in a concentration-dependent manner. Moreover, candesartan acyl-β-D-glucuronide inhibited the metabolism more strongly (IC50 = 18.9 µM) than did candesartan (IC50 = 150 µM) or candesartan N2-glucuronide (IC50 = 166 µM) (Fig. 5A; Table 1). Candesartan (IC50 > 250 µM), candesartan N2-glucuronide (IC50 = 191 µM), and candesartan acyl-β-D-glucuronide (IC50 = 159 µM) also inhibited the CYP3A4-mediated metabolism of paclitaxel in a concentration-dependent manner (Fig. 5B; Table 1).

Effects of candesartan and candesartan glucuronides on hydroxylation of paclitaxel by CYP2C8 (A) and CYP3A4 (B). CYP2C8 and CYP3A4 activities were determined by hydroxylation of paclitaxel to 6α-hydroxypaclitaxel and p-3′-hydroxypaclitaxel, respectively. Hydroxylation was determined by incubation of the recombinant proteins (CYP2C8, 10 pmol/ml; CYP3A4, 10 pmol/ml) and paclitaxel (2 and 4 µM, respectively) with the NADPH-regenerating system at 37°C for 30 minutes after a 5-minute preincubation. Each point represents the mean ± S.D. (n = 3).
Inhibition of CYP2C8 and CYP3A4 by candesartan and its glucuronides, as related to paclitaxel metabolism
Inhibition Models for Candesartan and Candesartan Acyl-β-D-Glucuronide.
The importance of TDI and mechanism-based inhibition of CYP for accurate estimation of drug-drug interaction has been recognized. As shown in Fig. 6, since preincubation with NADPH did not shift the IC50 values of candesartan acyl-β-D-glucuronide, the Ki values for CYP2C8-mediated hydroxylation of paclitaxel were determined by direct inhibition (no initial inhibition). It is clear from the Dixon plot that the inhibition of CYP2C8 by candesartan was noncompetitive (Ki = 133 ± 5.8 µM), whereas that by candesartan acyl-β-D-glucuronide was competitive (Ki = 7.12 ± 1.41 µM) (Fig. 7).

An IC50 shift assay for time-dependent inhibition of CYP2C8 by preincubation with NADPH and candesartan acyl-β-D-glucuronide. CYP2C8 activity was determined by hydroxylation of paclitaxel to 6α-hydroxypaclitaxel. Hydroxylation was determined by incubation of HLMs (0.1 mg protein/ml) with or without the initial addition of the NADPH-regenerating system, at 37°C for 30 minutes. The mixture was added to paclitaxel (2 µM) and incubated for 10 minutes. Each point represents the mean ± S.D. (n = 3).

Determination of Ki values for CYP2C8 inhibition by candesartan (A) and candesartan acyl-β-D-glucuronide (B). The activity of CYP2C8 was determined based on hydroxylation of paclitaxel to 6α-hydroxypaclitaxel. Hydroxylation was determined by incubation of recombinant CYP2C8 (10 pmol/ml), test compounds (candesartan, 15.6–250 µM; candesartan acyl-β-D-glucuronide, 3.8–60 µM), and paclitaxel (0.5, 1.0, and 2.0 µM) with the NADPH-regenerating system at 37°C for 30 minutes after a 5-minute preincubation. Each point represents the mean ± S.D. (n = 3).
Discussion
Candesartan, candesartan N2-glucuronide, and candesartan acyl-β-D-glucuronide inhibited the CYP2C8- and CYP3A4-mediated metabolism of paclitaxel with candesartan acyl-β-D-glucuronide exhibiting the strongest inhibition of CYP2C8-mediated paclitaxel metabolism without eliciting strong inhibition of CYP3A4 (Fig. 5; Table 1). It is therefore important to monitor the substrates of CYP2C8 rather than of CYP3A4 that are used for treating patients administered candesartan.
Gemfibrozil acyl glucuronide (Ogilvie et al., 2006) and clopidogrel acyl glucuronide (Tornio et al., 2014) strongly inhibit CYP2C8 in a time- and metabolism-dependent manner. However, although candesartan acyl-β-D-glucuronide strongly inhibited CYP2C8 (Fig. 5), the IC50 shift assay did not show preincubation time-dependent inhibition of CYP2C8 (Fig. 6). Therefore, the mechanism of CYP2C8 inhibition by candesartan acyl-β-D-glucuronide differs from that of gemfibrozil acyl glucuronide and clopidogrel acyl glucuronide.
Moreover, candesartan noncompetitively inhibited CYP2C8, whereas candesartan acyl-β-D-glucuronide showed a strong competitive inhibition (Fig. 7) that was not time-dependent (Fig. 6). Kumar et al. (2002) reported that hydroxylation of diclofenac acyl glucuronide to 4′-hydroxy diclofenac acyl glucuronide, which is a major pathway responsible for metabolizing 30% of the in vivo dose (Stierlin et al., 1979), is primarily catalyzed by CYP2C8 and not CYP3A4, CYP2C9, or CYP2D6 in the presence of NADPH and HLMs. Nishihara et al. (2012) revealed that sipoglitazar glucuronide becomes de-ethylated after incubation with HLMs, whereas CYP2C8 is predominantly involved in dealkylation. Although there are some reports that glucuronic acid conjugates serve as substrates for CYP2C8, incubation of 10 μM candesartan acyl-β-D-glucuronide with 10 pmol/ml CYP2C8 and the NADPH-regenerating system for 30 minutes did not induce degradation (Supplemental Fig. 1). It is necessary to consider factors other than the affinity of CYP2C8 for the substrate binding site as the mechanism by which candesartan acylglucuronide exhibits a CYP2C8 inhibitory effect. Jenkins et al. (2011) reported that multiple CYP2C8 inhibitors may contribute to the potent inhibitory effect of CYP2C8 but do not necessarily cause time-dependent inhibition by glucuronides, including gemfibrozil acyl glucuronide. In a comparative study using a docking simulator of certain gemfibrozil analogs in which the methyl group closest to heme was changed, they proposed that the presence or absence of a methyl group affects the time-dependent inhibition. Candesartan acyl-β-D-glucuronide contains a benzimidazole-7-carboxylate structure with glucuronized carboxylic acid. Candesartan cilexetil also contains a benzimidazole-7-carboxylate structure, and carboxylic acid is esterified with cilexetil (Fig. 1). Foti et al. (2016) reported that the degradation of candesartan cilexetil was minimal upon incubation with CYP2C8 with no appreciable formation of an ester-hydrolyzed product. Computational docking simulation showed that candesartan cilexetil strongly inhibited the CYP2C8-mediated amodiaquine metabolism (IC50 = 0.50 µM; Walsky et al., 2005) without interacting with the heme. Whether glucuronide conjugates can show more potent mechanism-based inhibition may not require that the glucuronides be a substrate for CYP2C8.
We show that not only UGT1A10 but also UGT2B7 produce candesartan acyl-β-D-glucuronide (Fig. 2). Most reports on candesartan metabolism do not mention glucuronidation as a metabolic pathway because in a phase I clinical trial in which candesartan cilexetil was administered to healthy subjects, only candesartan and its main oxidized metabolites were detected (Kondo et al., 1996). However, evidence for glucuronic acid metabolism in humans is indispensable for estimating the magnitude of drug-drug interactions between CYP2C8 substrates and candesartan acyl-β-D-glucuronide in clinical settings. Oral administration of 1 mg/kg candesartan cilexetil resulted in 0.8% of candesartan acyl-β-D-glucuronide excreted in feces 48 hours after administration. In the same study, administration of 1 mg/kg of candesartan cilexetil [C14] into the duodenum of rats through cannulated biliary tract resulted in excretion of candesartan acyl-β-D in feces having [C14] content that was 7.9% of the dose 24 hours after the administration. Hence, candesartan acyl-β-D-glucuronide likely undergoes deconjugation in the digestive tract and enters the enteric circulation as candesartan (Kondo et al., 1996), making it challenging to detect candesartan acyl-β-D-glucuronide in humans.
UGT1A10 mRNA is not detectable in the human liver; however, UGT1A10 is predominantly expressed in the intestine. UGT1A7, UGT1A8, and UGT1A9 mRNAs are expressed at lower levels than UGT1A10 in the intestine, whereas only UGT1A7 and UGT1A9 mRNAs are expressed in the liver (Ohno and Nakajin, 2009). Alonen et al. (2008) reported that UGT2B4, UGT2B10, UGT2B15, UGT2B17, and UGT2B28 do not contribute to the O-glucuronidation of candesartan. UGT2B7 mRNA is expressed at a 2-fold higher level in the intestine than UGT1A10 (Ohno and Nakajin, 2009). Thus, UGT2B7 must contribute to the formation of candesartan acyl-β-D-glucuronide in the liver. However, candesartan acyl-β-D-glucuronide formation is more complex in the liver than in the intestine. Interestingly, after intravenous administration of 14C-labeled candesartan N2-glucuronide, candesartan and candesartan acyl-β-D-glucuronide were detected in rat bile, whereas candesartan N2-glucuronide was not detectable (Kondo et al., 1996). Hence, candesartan acyl-β-D-glucuronide might be formed by transformation via acyl migration from N2-glucuronide to O-glucuronide or by enzymatic deconjugation via hydrolase and, subsequently, reglucuronization by UGT2B7. Further studies are warranted to clarify the mechanism of candesartan acyl-β-D-glucuronide formation in the liver.
The absolute bioavailability of candesartan after oral administration of candesartan cilexetil as a tablet or as an alcohol solution is 15% (Product Monograph; AstraZeneca, Canada) and 42.3% (Yamaoka et al., 1981), respectively. The low absorption of candesartan is likely caused by the efflux transporter P-glycoprotein as well as by its low solubility (Satturwar et al., 2007). In closed-loop pharmacokinetic studies with naringin, a P-glycoprotein substrate, using an in situ rat model, Surampalli et al. (2015) reported a 1.34-fold increase in area under the plasma concentration-time curve with an elevated peak concentration (Cmax) of candesartan. However, naringin is also a strong inhibitor of UGT1A10 (IC50 value = 3.4 µM) (Gufford et al., 2014). UGT-mediated glucuronidation is reportedly an important detoxification pathway in the intestine for numerous endobiotics or xenobiotics and is responsible for the first-pass pathway of several drugs, including raloxifene (Kemp et al., 2002), and phenolics (e.g., resveratrol, quercetin) (Wu et al., 2011). Cumulatively, these data may indicate that glucuronidation of candesartan in the intestine contributes to its low bioavailability.
We also identified candesartan glucuronides as substrates and moderate inhibitors of OATP1B1 and OATP1B3 (Fig. 3) compared with strong OATP1B1 inhibitors (i.e., cyclosporine, itraconazole, and rifampicin; Karlgren et al., 2012; McFeely et al., 2020). Specifically, at 10 µM, candesartan and acyl-β-D-glucuronide exhibited less than 50% inhibition of OATP1B1 and OATP1B3 (Fig. 4). Most drug interactions involving transporters depend on the unbound concentration of the drug. Indeed, 1 µM of candesartan was found to only slightly inhibit OATP1B1 and OATP1B3, with the estimated maximum total concentration ([I]inlet,max, 0.52 µM) and maximum unbound concentration of candesartan in the liver inlet ([I]u, inlet,max, 2.6 nM) calculated using the parameters [total hepatic blood flow rate = 97 l/h; unbound fraction (fu) = 0.005; Cmax = 0.22 µM (Hoogkamer et al., 1998); ka = 1.485 hour−1 (Meineke et al., 1997); dose = 12 mg of candesartan cilexetil; Fa × Fg = 1 (Ito et al., 1998)] not reaching the inhibitory concentration. Conversely, candesartan glucuronides were only detectable in the bile acid (Kondo et al., 1996), whereas their hepatic concentrations remain unknown. Therefore, at the clinical dose, candesartan and its glucuronides are not likely to reach sufficient concentrations to inhibit OATP1B1 and OATP1B3. Hence, there is a low probability of drug-drug interactions occurring involving OATP1B1 and OATP1B3 upon oral administration of candesartan cilexetil.
Our results also demonstrated that candesartan N2-glucuronide and candesartan acyl-β-D-glucuronide serve as substrates of OATP1B1 and OATP1B3 (Fig. 3), which are important transporters for hepatic uptake of various drugs from the portal and systemic circulating blood. Specifically, candesartan acyl-β-D-glucuronide exhibited high affinity for OATP1B3 compared with candesartan N2-glucuronide (Fig. 3). Several researchers have reported unique pharmacokinetics of glucuronides. For instance, gemfibrozil acyl glucuronide, a potent CYP2C8 inhibitor, is a substrate of OATP1B1, with a higher affinity than gemfibrozil (Shitara et al., 2004). Sallustio et al. (1996) reported that the liver:perfusate concentration ratio of gemfibrozil acyl glucuronide is 35:42. Vasilyeva et al. (2015) reported that sorafenib glucuronide was barely detectable in mouse plasma, the liver-to-plasma ratio of sorafenib glucuronide was approximately 350 after oral sorafenib administration, and the ratio markedly decreased in Oatp1a/1b knockout mice. Therefore, candesartan acyl-β-D-glucuronide may also accumulate at a high concentration in the liver. Kondo et al. (1996) further confirmed the enterohepatic circulation of candesartan using a radioisotope. Hence, after glucuronidation of candesartan in the intestine or hepatocytes, candesartan acyl-β-D-glucuronide might continuously accumulate in hepatocytes, being actively taken up by OATP1B1 and OATP1B3 and transported via the enterohepatic circulation, thereby facilitating its interaction with CYP2C8 substrates (Fig. 8).

Hypothesized mechanism for the potential drug interaction of candesartan cilexetil with paclitaxel. Hypothesized mechanism for the potential drug interaction of candesartan cilexetil with paclitaxel. Nearly all orally administered candesartan cilexetil becomes hydrolyzed to candesartan in the intestine, which is followed by glucuronidation to candesartan acyl-β-D-glucuronide by UGT1A10 and UGT2B7. Thereafter, candesartan acyl-β-D-glucuronide is actively transported into hepatocytes by OATP1B1 and OATP1B3. Candesartan is also transformed to acyl-β-D-glucuronide by UGT2B7 in the liver, and glucuronides are secreted in the bile and reabsorbed through enterohepatocyte circulation. This cycle may result in an interaction between a CYP2C8 substrate and candesartan acyl-β-D-glucuronide. Acyl glu, candesartan acyl-β-D-glucuronide; BCRP, breast cancer resistance protein; MRP, multidrug resistance protein; PTX, paclitaxel.
In summary, candesartan acyl-β-D-glucuronide is converted into a strong CYP2C8 inhibitor and functions as OATP1B1 and OATP1B3 substrate. Our findings suggest that after administration of candesartan cilexetil, candesartan acyl-β-D-glucuronide is actively transported by OATPs into hepatocytes, resulting in drug-drug interactions with CYP2C8 substrates, such as paclitaxel. These results provide new insights for considering the interaction between candesartan and paclitaxel in clinical practice that cannot be explained solely by inhibition of the paclitaxel excretion pathway by candesartan. However, owing to the large differences in the metabolic profiles of humans, rats, and dogs, it remains unclear whether glucuronic acid metabolites identified in in vivo studies are physiologically relevant to these various organisms. Therefore, further clinical studies on the interaction between CYP2C8 substrate drugs and candesartan as well as investigation into the metabolic profile of candesartan using human hepatocytes and small intestinal microsomes are needed.
Acknowledgments
We thank Hiroyuki Kusuhara and Kazuya Maeda (Laboratory of Molecular Pharmacokinetics, Tokyo University, Japan) for providing the HEK/OATP1B3 cells.
Authorship Contributions
Participated in research design: Katsube, Tsujimoto, Minegaki, Nishiguchi.
Conducted experiments: Katsube, Koide, Minegaki.
Performed data analysis: Katsube.
Wrote or contributed to the writing of the manuscript: Katsube, Tsujimoto, Koide, Hira, Ikeda, Minegaki, Morita, Terada, Nishiguchi.
Footnotes
- Received May 22, 2020.
- Accepted December 30, 2020.
↵1 Current affiliation: Department of Pharmacy, Kyushu University Hospital, Fukuoka, Japan.
↵2 Current affiliation: Department of Pharmacy, Shiga University of Medical Science Hospital, Shiga, Japan.
↵3 Current affiliation: Laboratory of Medicinal Cell Biology, Kobe Pharmaceutical University, Kobe, Japan.
All authors declare that no support, financial or otherwise, has been received from any organization that may have an interest in the submitted work, and there are no other relationships or activities that could appear to have influenced the submitted work.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CI
- confidence interval
- DCF
- 2′,7′-dichlorofluorescein
- DMEM
- Dulbecco’s modified Eagle’s medium
- FL
- fluorescein
- G6P
- glucose-6-phosphate
- G6PDH
- G6P dehydrogenase
- HBSS
- Hank’s balanced salt solution
- HEK
- human embryonic kidney
- HLM
- human liver microsome
- HPLC
- high-performance liquid chromatography
- OATP
- organic anion–transporting polypeptide
- TDI
- time-dependent inhibition
- UDPGA
- UDP–glucuronic acid
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}