


Abstract
Metabolism and disposition of pevonedistat, an investigational, first-in-class inhibitor of the NEDD8-activating enzyme (NAE), were characterized in patients with advanced solid tumors after intravenous infusion of [14C]pevonedistat at 25 mg/m2 (∼60–85 μCi radioactive dose). More than 94% of the administered dose was recovered, with ∼41% and ∼53% of drug-related material eliminated in urine and feces, respectively. The metabolite profiles of [14C]pevonedistat were established in plasma using an accelerator mass spectrometer and excreta with traditional radiometric analysis. In plasma, unchanged parent drug accounted for approximately 49% of the total drug-related material. Metabolites M1 and M2 were major (>10% of the total drug-related material) circulating metabolites and accounted for approximately 15% and 22% of the drug-related material, respectively. Unchanged [14C]pevonedistat accounted for approximately 4% and 17% of the dose in urine and feces, respectively. Oxidative metabolites M1, M2, and M3 appeared as the most abundant drug-related components in the excreta and represented approximately 27%, 26%, and 15% of the administered dose, respectively. Based on the unbound plasma exposure in cancer patients and in vitro NAE inhibition, the contribution of metabolites M1 and M2 to overall in vivo pharmacological activity is anticipated to be minimal. The exposure to these metabolites was higher at safe and well tolerated doses in rat and dog (the two preclinical species used in toxicology evaluation) plasma than that observed in human plasma. Reaction phenotyping studies revealed that CYP3A4/5 are primary enzymes responsible for the metabolic clearance of pevonedistat.
SIGNIFICANCE STATEMENT This study details the metabolism and clearance mechanisms of pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, after intravenous administration to patients with cancer. Pevonedistat is biotransformed to two major circulating metabolites with higher exposure in nonclinical toxicological species than in humans. The pharmacological activity contribution of these metabolites is minimal compared to the overall target pharmacological effect of pevonedistat. Renal clearance was not an important route of excretion of unchanged pevonedistat (∼4% of the dose).
Introduction
The ubiquitin-proteasome system (UPS) regulates normal cellular functions such as cell cycle, signal transduction, cell death, immune responses, and metabolism through intracellular protein degradation. This proteolytic process is catalyzed by a set of enzymes, ubiquitin-activating (E1) and ubiquitin-conjugating (E2) enzymes and ubiquitin ligases (E3) (Hershko and Ciechanover, 1998). These enzymes link chains of the polypeptide cofactor Ub onto proteins to mark them for degradation by the 26S proteasome (multicatalytic protease). Cullin-RING ligases (CRLs) are the largest family of ubiquitin E3 ligases that control turnover of many key substrates involved in cell cycle control, nuclear factor-kappa B (NF-κB) signaling, and DNA replication and repair (Zhao and Sun, 2013). CRLs are activated by covalent binding of ubiquitin-like protein NEDD8 (neural precursor cell-expressed, developmentally downregulated gene 8) to their cullin subunits through neddylation, which requires the E1 NEDD8-activating enzyme (NAE) as well as NEDD8-specific E2 and E3 enzymes. Deregulation of CRLs is linked to uncontrolled proliferative diseases such as cancer (Soucy et al., 2010). Thus, the NAE controls an early step in neddylation and represents a potential therapeutic target to modulate CRL activity and protein homeostasis (Zhao et al., 2014).
Pevonedistat [((1S,2S,4R)-4-{4-[(1S)-2,3-dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride; MLN4924; TAK-924) is a first-in-class, small-molecule, potent, and selective inhibitor of the NAE and is currently in clinical development across multiple cancers (Swords et al., 2018). Pevonedistat effectively blocks cullin neddylation to inhibit the activation of CRLs, leading to CRL substrate accumulation and the subsequent induction of multiple cell death pathways in cancer cells (Soucy et al., 2009). In xenograft models of diffuse large B-cell lymphoma (DLBCL) (OCI-Ly10 and OCI-Ly19) and acute myeloid leukemia (AML) (HL-60), pevonedistat demonstrated a dose-dependent decrease in neddylated cullins and a dose-dependent increase in CDL substrate pIκBα (phosphorylated protein inhibitor of nuclear [transcription] factor-κB) (Milhollen et al., 2010; Swords et al., 2010). Pevonedistat has been evaluated as a monotherapy in patients with a variety of advanced malignancies, including solid tumors (Sarantopoulos et al., 2016), relapsed or refractory lymphoma and multiple myeloma (Shah et al., 2016), and AML (Swords et al., 2015). It has also been evaluated in combination with azacitidine in AML (Swords et al., 2018) and with several chemotherapy regimens in patients with advanced solid tumors, including docetaxel and carboplatin plus paclitaxel (Lockhart et al., 2019). A phase 2 trial of pevonedistat combination therapy with azacitidine in patients with higher-risk myelodysplastic syndromes (MDSs), chronic myelomonocytic leukemia (CMML), and low-blast AML demonstrated encouraging clinical activity (NCT03814005) (Sekeres et al., 2021).
Pevonedistat was biotransformed via oxidative metabolism as a major clearance pathway in vitro and in preclinical species (rat and dog) (unpublished data). Metabolism and excretion studies in humans are routinely performed with radiolabeled investigational drug to determine circulating levels of a drug and its metabolites, to determine excretory metabolic pathways, and to elucidate the clearance mechanism of a drug (Penner et al., 2009; Coppola et al., 2019). Hence, an open-label phase 1 trial was conducted to evaluate the mass balance, metabolism, routes of elimination, and pharmacokinetics of [14C]pevonedistat after intravenous (i.v.) infusion to patients with advanced solid tumors (NCT03057366). The mass balance, pharmacokinetics, and excretion of [14C]pevonedistat have been reported recently (Zhou et al., 2021b). This article presents the characterization of circulating/excretory human metabolites and pharmacological activity of major circulating metabolites and assesses exposure coverage in preclinical toxicological species along with metabolic enzymes involved in the clearance of pevonedistat.
Materials and Methods
Test Articles and Reagents
[14C]Pevonedistat hydrochloride was synthesized at Quotient Sciences (Reading, UK). The radiochemical purity was ≥99% with high-performance liquid chromatography (HPLC) purity of 100%. The metabolite standard references M1 (ML00756900-001-C), M2 (ML00756899-001-B), and M3 (ML00756898-001-C) were synthesized at Millennium Pharmaceuticals, Inc. (Cambridge, MA), a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, with purity >98%. Ammonium acetate and acetic acid were purchased from Sigma-Aldrich (St. Louis, MO) and J.T. Baker (Phillipsburg, NJ), respectively. Liquid scintillation cocktail Ultima Gold and liquid cocktail MicroScint-PS for microplates (Isoplate-96) were obtained from PerkinElmer (Waltham, MA). The liquid scintillator FlowLogic U for beta-particle emission radioactivity monitor (β-RAM) was purchased from LabLogic Systems, Inc. (Brandon, FL). The human cDNA-expressed recombinant cytochrome P450s (rP450s) were purchased from Corning (Woburn, MA). Human liver S9 fractions (HS9) (mixed-gender pool of 200 donors) were purchased from Sekisui-Xenotech (Lenexa, KS). Acetonitrile (ACN), methanol, water, and all other reagents in high grade were purchased from commercial vendors.
Clinical Study Design and Dosing
Detailed clinical design, mass balance, and pharmacokinetics have been published recently (Zhou et al., 2021b). Briefly, the study was a single-center, open-label, nonrandomized study with two parts: part A was to evaluate the mass balance, metabolism, and excretion of pevonedistat after an intravenous infusion in patients with cancer, and part B (optional) was continued treatment with pevonedistat (20 mg/m2; days 1, 3, 5) in combination with chemotherapeutic agents (either docetaxel or carboplatin plus paclitaxel). This study was conducted in compliance with the independent ethics committee requirements of the participating region, informed consent regulations, and applicable regulatory requirements (including International Council for Harmonization guidelines) in accordance with ethical principles founded in the Declaration of Helsinki. The sterile liquid formulation (1 mg/ml) of pevonedistat was a solution containing citric acid (anhydrous) and beta cyclodextrin sulfobutyl ethers (Captisol), trisodium citrate dehydrate, and dextrose. [14C]Pevonedistat at 25 mg/m2 (approximately 80 μCi) was administered intravenously (60 minutes) to eight cancer patients with advanced solid tumors. The objective of this part of the study was to profile and identify circulating and excretory metabolites of pevonedistat. Blood samples were collected for measurement of total radioactivity (blood and plasma), pevonedistat concentration, and metabolite profiling at predose, end of infusion, and 0.5, 1, 2, 3, 4, 8, 12, 24, 48, 72, and every 24 hours thereafter until the release criteria were met. Urine samples (total radioactivity, pevonedistat, and metabolite profiling) were collected from 24 hours prior to dosing and at 0 to 6, 6 to 12, 12 to 24, and every 24 hours postdose until patients were released. Feces were collected at predose and at 24-hour intervals after dosing until release criteria were met and homogenized in water.
Sample Preparation for Metabolite Profiling
Plasma samples were pooled according to the Hamilton method across timepoints (0 to 72 hours) to create a single area under the curve (AUC)-representative pooled sample per patient (Hamilton et al., 1981). The pooling timepoints comprised >4 half-lives (t1/2) of the total radioactivity (TRA) in plasma. Pooled plasma (500 μl) samples were protein-precipitated with three volumes of acetonitrile, vortex-mixed for 5 minutes, stored at 4°C for 30 minutes, and centrifuged. Supernatant was transferred and reduced to partial dryness under a stream of nitrogen at 40°C until approximately 200 μl remained in the centrifuge tube. The concentrated extract was diluted with mobile phase A (10mM ammonium acetate, pH 5.6) and mobile phase B (ACN/methanol; 1:1, v/v), and recoveries (extraction and reconstitution) were assessed. Human urine and fecal homogenate samples were pooled by combining a constant percentage of volume/weight excreted during each collection interval and creating a single pooled sample per individual subject. The pooled timepoints were selected to achieve a minimum of ≥86% of the excreted radioactivity in urine or feces. Pooled urine samples were centrifuged, dried down under vacuum, and reconstituted with mobile phases A and B (1:1, v/v). Aliquots of pooled fecal homogenate samples were extracted three times with three volumes of ACN/methanol (1:1, v/v), and supernatants were separated. The combined extracts were dried under vacuum and reconstituted with mobile phases A and B (1:1, v/v). The extraction, reconstitution, and column recovery were assessed for individual subjects. Metabolite profiling of urine and fecal homogenate extracts was performed using conventional HPLC. Radioactivity detection was performed using a β-RAM radioactivity detector, whereas that of plasma extracts was performed using HPLC fractionation and accelerator mass spectrometer (AMS).
HPLC Conditions and Excreta Profiling
The chromatographic separations were achieved on a Phenomenex Luna C18 (2) column (250 × 4.6 mm, 5 μm, 100Å) (Phenomenex, Inc., Torrance, CA) with a Phenomenex guard column cartridge (Phenomenex, Inc.) at 40°C on an Agilent 1290 Infinity HPLC system (Agilent Technologies, Wilmington, DE). Mobile Phases A and B were 10 mM ammonium acetate (pH 5.6) and ACN/methanol (1:1, v/v), respectively. The flow rate was 0.7 ml/min with a linear gradient from 5%B to 10%B in 5 minutes, followed by a ramp to 40%B in 20 minutes. It then went to 30%B in 30 minutes, back to 40%B in 35 minutes, to 60%B in 40 minutes, a ramp to 100%B until 50 minutes with hold up to 55 minutes, and back to 5%B at 60 minutes. The UV detector was set at 276 nm. A 500-μl liquid cell (LabLogic Systems Inc.) was used for the β-RAM radiometric detector, and the flow rate of liquid scintillator FlowLogic U (LabLogic System Inc.) was 1.5 ml/min. For urine samples, HPLC elutes were collected in OptiPlate-96 (white opaque 96-well microplate) by the fraction collector, dried, and counted for 5 to 10 minutes by microplate counter (MicroBeta2; PerkinElmer) by mixing with scintillation cocktail. The system operation was controlled by Laura software Version 4.2.4.50 (LabLogic Systems Inc.). The radiochromatograms were integrated by Laura to obtain the percent distributions for the metabolites as shown by the radio-peaks. Fecal metabolite profiles were generated using the online radioactivity detector β-RAM Model 5C (LabLogic Systems Inc.).
Plasma Metabolite Profiling with AMS
The t1/2 of plasma total radioactivity is approximately 15 hours; hence, plasma was pooled up to 72 hours to reflect four to five half-lives of total radioactivity. Due to pooling of plasma up to 72 hours, the radioactivity concentration in the pooled sample was deemed insufficient for detection by traditional radioactivity detection methods (e.g., using β-RAM); thus, AMS was used for plasma metabolite profiling. The HPLC analysis was performed using the method described in the previous section, and eluants were collected as a series of fractions every 15 seconds. An equal proportion of volume from each individual fraction was pooled for AMS analysis. The pooled samples were dried under vacuum in the presence of copper oxide and placed into a combustion tube, which was heat-sealed under vacuum and heated at 900°C for 1 hour in a chamber furnace (Keck et al., 2010; Tse et al., 2014). The carbon dioxide generated by combustion was cryogenically transferred to a tube containing zinc powder and titanium hydride and graphitized by placing in a furnace and heating at 550°C for 5 hours. The graphite pellet was prepared from graphitized material and loaded onto the sample wheel, which was inserted into the ion source of the AMS instrument. The ion source (multi-cathode source of negative ions by cesium sputtering) created a beam of negative ions by directing the Cs ions on to the graphite sample and sputtering the surface. After sequential injection of 12C-, 13C-, and 14C- into the accelerator that accelerates and converts into positive ions, the abundant 12C+ and 13C+ ions were collected in the off-axis Faraday cups, and their respective currents were measured very precisely using a current integrator. The 14C+ ions were selected based on their charge state using an electrostatic analyzer and counted using a silicon surface barrier detector. The AMS measured the [14C]:[12C] ratio derived from all sources of carbon in the sample. The AMS results were expressed as percent Modern Carbon (pMC), where 100 pMC equals 13.56 dpm/g C, and plotted with Laura software; relative percentages were calculated. Regions of radioactivity were identified using a UV chromatogram of plasma extract spiked with unlabeled pevonedistat, M1, M2, and M3 reference standards. Metabolites M7-, M10b-, M16-, and M22-related regions were identified based on the known retention times from urine sample analysis and mass spectrometry of plasma samples.
Metabolite Identification
Metabolites in urine, fecal homogenates, and plasma samples were identified using a Triple TOF 5600 high-resolution mass spectrometer (AB SCIEX, Framingham, MA) in positive ionization modes. The HPLC eluate was introduced via an electrospray positive ionization source directly into the mass spectrometer with the following conditions: collision energy (CE) = 45 ± 15 V; curtain gas = 30 (arbitrary units); declustering potential = 60; and source temperature = 450°C. The mass defect tolerance was set at 50 with the parent’s mass. The data were acquired using Analyst TF 1.7 software (AB SCIEX) and processed with PeakView 2.1 software (AB SCIEX). Putative structures of metabolites were determined using elemental composition derived from the accurate mass, change in elemental composition relative to the parent drug, retention time when reference standards were available, and tandem mass spectrometry (MS/MS) fragmentation.
P450 Reaction Phenotyping of Pevonedistat
To determine the in vitro intrinsic clearance of pevonedistat in rP450s, pevonedistat (0.1 μM) was incubated with rP450s 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2J2, 3A4, and 3A5 (20 pmol/ml). The total protein concentration was adjusted to 0.5 mg/ml (except for CYP1A1 to 0.56 mg/ml) with appropriate control Supersomes (Corning) showing no endogenous P450 activity (control Supersomes with either P450 oxidoreductase or with both P450 oxidoreductase and cytochrome b5). The incubations were conducted in triplicate in 0.1 M potassium phosphate buffer supplemented with 2 mM nicotinamide adenine dinucleotide phosphate (NADPH) and 3 mM magnesium chloride (pH 7.4, 37°C) over 0, 3, 7, 12, 20, and 30 minutes. The reactions were terminated by the addition of 0.1 μM d8-pevonedisat (internal standard) in ACN. A 50-μl aliquot of the supernatant was then further diluted with 100 μl of water prior to analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS). To evaluate the role of the CYP4F family in pevonedistat metabolism, pevonedistat (0.1 μM) was also incubated with CYP4F2, 4F3A, 4F3B, and 4F12 as above, except only for 0- and 30-minute time points. The disappearance of pevonedistat over time was monitored by LC-MS/MS. Relative activity factor for each P450-enzyme was established using P450-specific probe substrate for each P450: 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5. Intrinsic clearance (CLint) in recombinant P450s (CLint,rP450) was calculated for pevonedistat and extrapolated to human liver microsomal (HLM) clearance for the P450 of interest using the relative activity factor approach (Venkatakrishnan et al., 2000, 2001). The relative contribution of each hepatic enzyme toward the metabolic clearance of pevonedistat was assessed.
Intrinsic clearance (CLint) of pevonedistat at 0.1 μM concentration was determined in human S9 fraction (2.5 mg/ml) in the presence of NADPH (30-minute incubation) and with and without the CYP3A-specific inhibitor ketoconazole (0.5 μM). The percent inhibition of the metabolism of pevonedistat by ketoconazole was assessed from this data.
Pharmacological Activity of Metabolites
In vitro pharmacological activity of pevonedistat and major circulating metabolites M1 and M2, and along with metabolite M3, was evaluated in: 1) a biochemical assay for NAE inhibition; 2) a cell-based NF-κB-luciferase reporter assay in human embryonic kidney (HEK)293 cells in which the luciferase signal requires NAE-dependent ubiquitination of the NF-κB inhibitor IκBα; and 3) a tumor cell viability assay in HCT116 (human colorectal carcinoma cell line) cells as per published procedure (Soucy et al., 2009).
Metabolite Profiling of Rat and Dog Plasma
Male Sprague-Dawley rats were administered 30 mg (100 μCi)/kg of [14C]pevonedistat. Thirty milligrams per kilogram was a safe and tolerable dose in toxicology studies in the rat. Terminal blood samples from rats were collected at 0.25, 0.5, 0.583, 0.833, 1.5, 3.5, 6.5, 8.5, 12.5, 24.5, 48.5, 72.5, 96.5, 120.5, 144.5, and 168.5 hours from the start of infusion (three rats/timepoint). Predose and individual plasma samples were pooled across timepoints and animals using the trapezoidal AUC pooling method (Hamilton et al., 1981). The drug-derived radioactivity from pooled plasma was extracted twice from plasma samples using 2× v/v of acetonitrile. The metabolite profiles were established using HPLC fraction collection and detection of radioactivity by Packard TopCount NXT Microplate Scintillation and Luminescence Counter technology.
Three male beagle dogs received a single i.v. infusion (30 minutes) of [14C]pevonedistat at 15 mg (40 μCi)/kg. Fifteen milligrams per kilogram was a safe and tolerable dose in toxicology studies in the dogs. Blood samples were collected from all dogs prior to dosing and at approximately 0.5 (end of infusion), 0.75, 1, 3, 6, 8, 10, 24, 48, and 72 hours after start of infusion. Predose and individual plasma samples were pooled across timepoints using the trapezoidal AUC pooling method. Aliquots of pooled plasma samples were extracted with ACN (1:3, v/v) three times and processed for metabolite profiling by HPLC/β-RAM or HPLC-fraction collector followed by a microplate reader. All the procedures and protocols for in vivo animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health and were approved by the Institution’s Animal Care and Use Committee or local equivalent.
Data Processing and Analysis
Metabolite profiles were generated by directly acquiring the data from β-RAM or importing MicroBeta/AMS data using the liquid scintillation counting import function in Laura software (LabLogic Systems, Inc.). Percent distribution of pevonedistat and metabolites was calculated by direct integration of the peak areas from metabolite profiles and converted to percent dose of metabolites.
The measured CLint with individual cytochrome P450s (P450s) was extrapolated to HLM clearance for the P450 of interest (P450i) using the relative activity factor (RAF) for P450i as shown in eq. 1:
 where CLint, HLM,P450i = intrinsic clearance in HLM by P450i (ml/min/mg protein); CLint,rP450i = intrinsic clearance by rP450i (ml/min/pmol P450); and RAF (determined using P450-specific probe substrate) = CLint,HLM,P450i/CLint,rP450i (pmol P450/mg protein).
where CLint, HLM,P450i = intrinsic clearance in HLM by P450i (ml/min/mg protein); CLint,rP450i = intrinsic clearance by rP450i (ml/min/pmol P450); and RAF (determined using P450-specific probe substrate) = CLint,HLM,P450i/CLint,rP450i (pmol P450/mg protein).
The percent contribution of individual hepatic P450 was then determined using eq. 2:
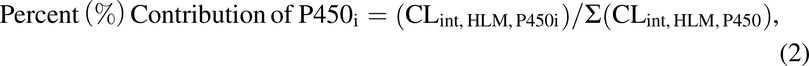 where Σ(CLint,HLM,P450) = sum of intrinsic clearance by P450s 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5.
where Σ(CLint,HLM,P450) = sum of intrinsic clearance by P450s 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5.
Results
Excretion of Drug-Related Material
Eight cancer patients with advanced solid tumors each received a 25-mg/m2 (∼60–85 μCi) single i.v. infusion of [14C]pevonedistat for 60 minutes in part A of the study (Zhou et al., 2021b). Although metabolite profiling was performed for all eight patients who completed part A of the study, the results of one patient were excluded in the final summary due to use of CYP3A-inducing medication (carbamazepine) that was included in the protocol’s exclusion list. The mean recovery of radioactivity in urine and feces combined was approximately 94% by 1 week post–intravenous administration of [14C]pevonedistat. Most of the radioactivity was recovered by 96 hours postdose. A mean of 53% and 41% of the dose was recovered in feces and urine, respectively (Zhou et al., 2021b). The mean t1/2 of pevonedistat and drug-related material (total radioactivity) in plasma was 8.4 hours and 15.6 hours, respectively. The unchanged parent accounted for approximately 41% of AUClast (area under the concentration-time curve until last sampling time) of total radioactivity, suggesting that 59% of the circulating radioactivity was due to metabolites (Zhou et al., 2021b).
Metabolite Profiles in Plasma
Metabolite profiles of AUC-pooled plasma (0 to 72 hours) for individual subjects were established using HPLC fractionation followed by AMS analysis. The mean samples processing (extraction and reconstitution) and column recoveries were determined as 90% and 106%, respectively. Percent distribution of pevonedistat and its metabolites in plasma are summarized in Table 1, and the structure of metabolites are presented in Fig. 1. The reconstructed HPLC radiochromatograms showed the presence of six circulating metabolites (Fig. 2A). The most abundant radiopeak was due to unchanged pevonedistat contributing to approximately 49% of the total circulating radioactivity. This is close to the ratio (∼41%) obtained from the AUC of the parent drug from direct measurement of the parent drug concentration using a validated bioanalytical method and to that of the total radioactivity calculated from the time-concentration profile. Hydroxylated metabolites M1 and, further oxidized to a ketone metabolite, M2 appeared as the major (>10% of the TRA) metabolites in plasma; these represented approximately 15% and 22% of the total radioactivity, respectively (Table 1). Metabolites M3 (hydroxylation) and M10b (oxidation and glucuronidation) accounted for approximately 5% and 7% of the total radioactivity, respectively. Metabolites M7 (N-dealkylation), M16 (direct glucuronidation), and M22 (oxidation and glucuronidation) were relatively minor metabolites, and each represented ≤2% of the total radioactivity (Table 1). The structure of minor metabolite M23 could not be identified.

Biotransformation pathways of pevonedistat in humans. *Position of 14C-label. FE, feces; Gluc., glucuronide; PL, plasma; UR, urine.

Representative radiochromatograms of (A) plasma, (B) urine, and (C) feces after intravenous infusion of [14C]pevonedistat. Pevo, [14C]pevonedistat.
Relative distribution of metabolites in pooled plasma (% drug-related material), urine (% dose), and feces (% dose) after intravenous administration of [14C]pevonedistat Metabolite profiles were presented for n = 7
Metabolite Profiles in Excreta
The percentages of the dose excreted as pevonedistat and metabolites are summarized in Table 1. The mean samples processing (urine: 102%; feces: 97%) and column (urine: 104%; feces: 98%) recoveries were >95%. Metabolite profiles of pooled urine samples revealed the presence of up to nine metabolites (Fig. 2B). Approximately 41% of the administered dose was excreted in urine. Unchanged pevonedistat accounted for approximately 4% of the administered dose in urine. M1 was the major metabolite, which represented approximately 20% of the administered dose. Metabolites M2, M3, and M16 represented approximately 8%, 4%, and 3% of the dose, respectively. M7, M10b, and M22 are minor metabolites, and each accounted for <2% of the administered dose. Approximately 53% of the administered dose was excreted in feces. Unchanged pevonedistat accounted for approximately 17% of the dose (Fig. 2C; Table 1). M1, M2, and M3 were major metabolites that accounted for approximately 7%, 18%, and 11% of the dose, respectively. Metabolite M7 was a minor metabolite and accounted for <1% of the dose.
Overall, ∼74% of the dose was excreted as metabolites in urine and feces, combined with 21% (4% and 17% of the dose in urine and feces, respectively) as parent drug.
Identification of Metabolites
The structures of the metabolites were characterized by measuring the change in both elemental composition and characteristic fragment ions and their accurate mass-to-charge ratio (m/z) compared with that of pevonedistat (Fig. 1; Table 2). The biotransformation of pevonedistat to metabolites M1, M2, and M3 resulted from hydroxylation (M1 and M3, +16 Da) and subsequent oxidation to a ketone (M2, +14 Da). Additionally, the identities of metabolites M1, M2, and M3 were confirmed by comparison with the retention time of the corresponding reference standards (Table 2).
Pevonedistat metabolites detected and identified in plasma, urine, and feces of patients administered an intravenous dose of pevonedistat
P450 Reaction Phenotyping
The metabolite profiling data from this study confirmed that pevonedistat was primarily cleared via metabolism, with oxidation being the predominant pathway. Metabolites M1, M2, and M3 accounted for approximately 68% of the total dose excreted in urine and feces combined (Table 1). All oxidative pathways combined represented ∼72% of the administered dose. To determine the enzymes responsible for pevonedistat metabolism, the clearance of pevonedistat was measured in the presence of rP450s: CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, 4F2, 4F3A, 4F3B, and 4F12. Only CYP3A4/5, 2C8, 2J2, and 1A1 were shown to meaningfully contribute to the pevonedistat metabolism with CLint,rP450 3.27E-03, 1.04E-04, 7.01E-04, and 7.01E-04 ml/min/pmol P450, respectively (Table 3). Since CYP1A1 and 2J2 are primarily extrahepatic enzymes with low expression in the liver, the quantitative contribution of these P450s could not be assessed. The quantitative contribution of P450s in the clearance of pevonedistat was assessed for the following hepatic P450s only: CYP1A2, 2B6, 2C8, 2C9, 2D6, and 3A4/5 using the RAF. Among hepatic P450s, CYP3A4/5 contributed approximately 94% to the overall hepatic metabolism, and CYP1A2, 2B6, 2C8, 2C9, 2C19, and 2D6 contributed <2% each (Table 3).
Intrinsic clearance of pevonedistat (0.1 μM) by recombinant cytochrome P450 isozymes
Pharmacological Activity of Metabolites
In vitro pharmacological activity of pevonedistat and its oxidative metabolites (M1, M2, and M3) was assessed in biochemical, reporter, and cell viability assays; the results are summarized in Table 4. Pevonedistat and its oxidative metabolites—M1, M2, and M3—exhibited similar 50% inhibitory concentration (IC50) values (0.004–0.010) compared with pevonedistat (within 2-fold range) in an NAE inhibition biochemical assay. However, in the cell-based NF-κB inhibition assay (the most relevant assay for target pharmacological activity), M1, M2, and M3 were 8- to 13-fold less potent than pevonedistat (IC50 [μM]): pevonedistat: 0.016; M1: 0.13; M2: 0.13; M3: 0.20, and in the HCT116 viability assay (dose-causing lethality for 50% of the population [LD50] [μM]: pevonedistat: 0.12; M1: 0.41; M2: 0.43; M3: 0.99), the metabolites were also less potent, in the 3- to 8-fold range (Table 4). Based on the plasma exposure, unbound fraction in plasma, and NF-κB inhibition IC50, the contribution of metabolites to the overall pharmacology of pevonedistat is expected to be minor.
Pharmacological activity of pevonedistat and its metabolites: M1, M2, and M3
Cross-Species Comparison of Metabolites
The excretion, mass balance, and metabolism of pevonedistat were also evaluated after intravenous administration of 14C-pevonedistat in rats and dogs, the two species used in the toxicology assessment for pevonedistat (data on file). In general, the exposure to the circulating metabolites in toxicological species at doses that were safe and well tolerated are broadly similar to or higher than that observed in humans. There were no human-specific circulating metabolites identified in this study. A quantitative comparison of the two major metabolites (>10% of TRA in human plasma) M1 and M2 in rats, dogs, and humans is presented in Table 5. M1 and M2 represented approximately 15% and 22% of the drug-related material in human plasma, which is equivalent to 545 and 802 hour*ng*Eq/ml, respectively. Both metabolites M1 and M2 were present at approximately 1- to 4-fold and 2- to 6-fold higher exposure in rat and dog plasma, respectively.
Exposure of M1 and M2 in rat, dog, and human plasma
Discussion
Pevonedistat is a first-in-class inhibitor of the NAE and is currently in clinical development across multiple cancers, including low-blast AML and high-risk myelodysplastic syndrome (MDS) (NCT03814005). A human metabolism and excretion study with [14C]pevonedistat was conducted to understand the mass balance and routes of elimination and to elucidate clearance mechanisms of pevonedistat in patients with advanced cancer (Zhou et al., 2021b). The quantitative information on circulating and excretory metabolites obtained from the absorption, distribution, metabolism, and excretion (ADME) study is critical to understand the potential for drug-drug interactions (DDIs) as a victim and the contribution of metabolites to the pharmacology/toxicology of a parent drug. In addition, information on the level of circulating metabolites is important to assess DDI potential for metabolites as perpetrators. The mean total recovery in urine and feces was greater than 90% of the administered radioactivity. Most of the dose was excreted within 4 days of pevonedistat administration, and excretion was essentially complete in 1 week. The excretion profiles were similar across subjects with low intersubject variability. Total drug-related material in plasma declined at a slightly slower rate than pevonedistat, suggesting that some metabolites have longer half-lives than the parent drug (Zhou et al., 2021b).
If designed and conducted appropriately, the radiolabeled human metabolism and disposition study is one of the key clinical pharmacology studies that can provide a rich source of information related to absorption, metabolism, and excretion of a new chemical entity (Penner et al., 2009; Spracklin et al., 2020; Rowland Yeo and Venkatakrishnan, 2021). Recently, Coppola et al. (2019) reviewed the marketing authorization applications submitted during last the 10 years to the European Medicines Agency and highlighted many shortcomings related to the characterization of circulating and excretory metabolites from human ADME studies.
Despite extensive literature and guidance, inadequate sample pooling and characterization of circulating metabolites in plasma were observed in many cases. In the current study, for metabolite profiling, plasma samples were pooled for up to 72 hours using the AUC pooling method to capture approximately four to five half-lives of drug-related material (Hamilton et al., 1981). Due to pooling of up to 72 hours, plasma radioactivity was diluted and metabolite profiling could not be performed using traditional radioactivity detection methods. This is one of the limitations of AUC pooling methods; accordingly, an ultrahighly sensitive AMS method was used to detect low amounts of radioactivity in the metabolite profiles of the plasma pools. The unchanged pevonedistat is the most abundant (∼49%) radioactivity-related peak (Fig. 2A; Table 1) in plasma. This is close to the AUClast (area under the concentration-time curve until last sampling time) ratio (∼41%) for pevonedistat to total radioactivity measured with the validated LC-MS/MS method. The minor difference in pevonedistat levels may be due to differences in time intervals assessed (48 hours vs. 72 hours) and/or differences in sensitivity and/or extraction efficiencies between AMS and LC-MS/MS methods. M1 and M2 each represented >10% of the total circulating drug-related material. Other metabolites—M3, M10b, M7, M16, and M22—were minor, as each represented <10% of total radioactivity.
Pevonedistat was primarily metabolized via hydroxylation and oxidation. M1 and M3 are hydroxylated metabolites (epimeric) that accounted for ∼27% and ∼15% of the administered dose, respectively, excreted in urine and feces combined. M2 is the result of further oxidation of M1 and/or M3 and accounted for 27% of the administered dose. After intravenous administration, ∼4% and ∼17% of the dose were recovered as unchanged parent drug in urine and feces, respectively. The unchanged parent drug in feces may have resulted from either biliary excretion or intestinal secretion after intravenous administration, although according to preclinical studies, intestinal secretion does not appear to be a contributor (unpublished results). M1 and M3 are epimeric metabolites, and their formation is primarily catalyzed by CYP3A4/5 enzymes. Metabolite M2 could be formed by P450 enzymes and/or alcohol dehydrogenases mediated oxidation of M1 and M3, and this process is documented in the literature (Diao et al., 2013; Di et al., 2021). Conversely, M2 could also be converted back to M1 and M3 by aldo-keto reductases and/or the short-chain dehydrogenases/reductases (e.g., carbonyl reductase) (Barski et al., 2008; Shi and Di, 2017). However, the mechanism and enzymes associated with the formation of M2 and its reduction back to M1 and M3 were not determined in this study but may be a subject of future investigations. The excretion of a very low level of unchanged pevonedistat in urine suggested that the renal clearance was not an important route of excretion of pevonedistat after intravenous administration.
The human metabolism and excretion study and the P450 phenotyping study revealed that oxidative metabolism was the predominant metabolic pathway of pevonedistat and was primarily catalyzed by CYP3A4/5 (approximately 94% contribution). In liver microsomes, 88% of intrinsic clearance of pevonedistat was inhibited by ketoconazole (0.5 μM), a CYP3A4/5 inhibitor, consistent with the data from reaction phenotyping experiments with rP450s. The metabolite profiling data from human excreta indicated that at least 74% of the administered dose was cleared via oxidative metabolism. Thus, the data from in vitro experiments are consistent with data observed in vivo in the human metabolism and excretion study. Although CYP3A4/5 were the primary enzymes responsible for the metabolic clearance of pevonedistat, a clinical DDI study with a strong CYP3A4/P-glycoprotein inhibitor, itraconazole, did not show any meaningful increase in pevonedistat systemic exposure (Faessel et al., 2019). Hepatic uptake parameters were assessed in sandwich-cultured hepatocytes (unpublished results). Development of a physiologically based pharmacokinetic model by incorporation of hepatic uptake parameters (CLPD, Jmax, Km) indicated that the systemic exposure of the drug is not sensitive to the perturbations of hepatic enzyme activity in the liver when the hepatic uptake becomes the rate-determining step of drug clearance. The results of mechanistic investigations, including physiologically based pharmacokinetic modeling exploring the lack of DDI for pevonedistat with CYP3A4/P-glycoprotein inhibitors and inducers, have been recently presented (Zhou et al., 2021a). This phenomenon has been described for other drugs as reported in the literature (Maeda et al., 2011; Yoshikado et al., 2017, Patilea-Vrana and Unadkat, 2018).
NAE is an essential E1 enzyme that initiates a protein homeostatic pathway essential for cancer cell growth and survival through the activation of a family of E3 ubiquitin ligases known as CRLs. When NAE is functional, CRL substrates involved in processes including DNA replication, cell cycle regulation, and NF-κB signaling are ubiquitinated and degraded in a regulated manner. In contrast, inhibition of NAE with pevonedistat results in accumulation of these CRL substrates and can lead to effects including DNA rereplication and inhibition of NF-κB (Soucy et al., 2010). The pharmacological activity of pevonedistat and metabolites M1–M3 was assessed in biochemical, reporter, and cell viability assays. Although pharmacological activity of the major circulating metabolites M1 and M2 was similar to pevonedistat in a cell-free biochemical assay of NAE activity, when evaluated in cell-based assays, the activity of the metabolites was much weaker than that of pevonedistat. The shift in IC50 values (3- to 13-fold) from biochemical to pharmacologically more relevant cellular assays is likely due to differences in cellular permeability between pevonedistat and the metabolites. By including relative concentration, the plasma unbound fraction, and differences in cellular potency (NF-κB inhibition) of pevonedistat and metabolites, the contributions of metabolites to overall pharmacology activity were found to be minimal and are not expected to be clinically relevant.
After the administration of pevonedistat, metabolites M1 and M2 exceeded the 10% threshold of circulating metabolites as described in the US Food and Drug Administration Safety Testing for Drug Metabolites Guidance and the International Council on Harmonisation M3 (R2) documents (European Medicines Agency, 2008; US Department of Health and Human Services, 2020). The exposure of these two metabolites was assessed in the rat and dog ADME studies at doses that were safe and well tolerated (Table 5). Both M1 and M2 were major circulating metabolites in rats and dogs and were present at approximately 1- to 6-fold higher concentration in animals relative to that in humans, suggesting that adequate animal exposure was achieved in the toxicological species in safety assessment studies.
In summary, the biotransformation and clearance pathways of pevonedistat were characterized after i.v. infusion of 25 mg/m2 to patients with cancer. The mass balance results confirmed that drug-related material was excreted via both hepatic and renal elimination routes. However, renal elimination of unchanged pevonedistat was minor. Approximately 74% of the administered dose was recovered as oxidative metabolites in the excreta. The contribution of major circulating metabolites M1 and M2 to overall pharmacological activity of pevonedistat is minimal.
Acknowledgments
The authors acknowledge Pharmaron for plasma metabolite profiling assistance using AMS analysis. Editorial support, conducted in accordance with Good Publication Practice 3 and the International Committee of Medical Journal Editors guidelines, was provided by Jacquelyn Williams, MSc, of Oxford PharmaGenesis, Inc. (Newtown, PA) and funded by Takeda Pharmaceuticals U.S.A., Inc.
Authorship Contributions
Participated in research design: Bolleddula, Chen, Cohen, Zhou, Pusalkar, Sedarati, Venkatakrishnan, Chowdhury.
Conducted experiments: Chen, Cohen.
Performed data analysis: Bolleddula, Chen, Cohen, Pusalkar, Berger, Chowdhury.
Wrote or contributed to the writing of the manuscript: Bolleddula, Chowdhury.
Footnotes
- Received January 27, 2022.
- Accepted March 29, 2022.
This work was supported by Millennium Pharmaceuticals, Inc. (Cambridge, MA), a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. This work received no external funding.
All authors are current or former employees of Takeda. A.B. declares stock or stock options in Takeda. All other authors declare no competing interests.
↵1 Current affiliation: EMD Serono Research & Development Institute, Billerica, Massachusetts.
↵2 Current affiliation: Servier Pharmaceuticals, Boston, Massachusetts.
3 Current affiliation: Boston Pharmaceuticals, Cambridge, Massachusetts.
Abbreviations
- ACN
- acetonitrile
- ADME
- absorption, distribution, metabolism, and excretion
- AML
- acute myeloid leukemia
- AMS
- accelerator mass spectrometer
- AUC
- area under the curve
- CLint
- intrinsic clearance
- CLint,rP450
- intrinsic clearance in recombinant P450
- CRL
- cullin-RING ligase
- DDI
- drug-drug interaction
- E1
- ubiquitin-activating enzyme
- E3
- ubiquitin ligase
- HPLC
- high-performance liquid chromatography
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- m/z
- mass-to-charge ratio
- NAE
- NEDD8-activating enzyme
- NEDD8
- neural precursor cell-expressed, developmentally downregulated gene 8
- NF-κB
- nuclear factor-kappa B
- P450
- cytochrome P450
- P450i
- P450 of interest
- RAF
- relative activity factor
- β-RAM
- beta-particle emission radioactivity monitor
- rP450
- recombinant cytochrome P450
- t1/2
- half-life
- TRA
- total radioactivity
- Copyright © 2022 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}