


Visual Overview

Abstract
Drug metabolite identification is an integrated part of drug metabolism and pharmacokinetics studies in drug discovery and development. Definitive identification of metabolic modification sides of test compounds such as screening metabolic soft spots and supporting metabolite synthesis are often required. Currently, liquid chromatography-high resolution mass spectrometry is the dominant analytical platform for metabolite identification. However, the interpretation of product ion spectra generated by commonly used collision-induced disassociation (CID) and higher-energy collisional dissociation (HCD) often fails to identify locations of metabolic modifications, especially glucuronidation. Recently, a ZenoTOF 7600 mass spectrometer equipped with electron-activated dissociation (EAD-HRMS) was introduced. The primary objective of this study was to apply EAD-HRMS to identify metabolism sites of vepdegestrant (ARV-471), a model compound that consists of multiple functional groups. ARV-471 was incubated in dog liver microsomes and 12 phase I metabolites and glucuronides were detected. EAD generated unique product ions via orthogonal fragmentation, which allowed for accurately determining the metabolism sites of ARV-471, including phenol glucuronidation, piperazine N-dealkylation, glutarimide hydrolysis, piperidine oxidation, and piperidine lactam formation. In contrast, CID and HCD spectral interpretation failed to identify modification sites of three O-glucuronides and three phase I metabolites. The results demonstrated that EAD has significant advantages over CID and HCD in definitive structural elucidation of glucuronides and phase I metabolites although the utility of EAD-HRMS in identifying various types of drug metabolites remains to be further evaluated.
SIGNIFICANCE STATEMENT Definitive identification of metabolic modification sites by liquid chromatography-high resolution mass spectrometry is highly needed in drug metabolism research, such as screening metabolic soft spots and supporting metabolite synthesis. However, commonly used collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) fragmentation techniques often fail to provide critical information for definitive structural elucidation. In this study, the electron-activated dissociation (EAD) was applied to identifying glucuronidation and oxidative metabolism sites of vepdegestrant, which generated significantly better results than CID and HCD.
Introduction
Drug metabolite identification is an integrated part of drug metabolism and pharmacokinetics studies in the drug discovery and development process, which provides insights into its pharmacokinetics, pharmacodynamics, and toxicity. Currently, liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS) is the dominant analytical platform for drug metabolite profiling and identification. In the LC-HRMS analysis, the first step is the acquisition of accurate mass liquid chromatography-mass spectrometry (LC-MS) and liquid chromatography tandem mass spectrometry (LC-MS/MS) datasets of a test compound and its metabolites in biologic samples, which is routinely carried out using data-dependent acquisition (DDA) methods such as ion intensity, mass defect (Zhang et al., 2009), pseudo-neutral loss (Castro-Perez et al., 2005), and background exclusion (Comstock et al., 2019). As an alternative, data-independent acquisition methods, such as MSE (Wrona et al., 2005; Bateman et al., 2007; Tiller et al., 2008; Bonn et al., 2010) and sequential windowed acquisition of all theoretical fragment ions (Gillet et al., 2012; Hopfgartner et al., 2012; Collins et al., 2017; Ludwig et al., 2018) have been used for fast drug metabolite profiling. The second step is the data processing to find metabolite ions in recorded LC-MS and LC-MS/MS data using targeted extracted ion chromatography (Ma and Chowdhury, 2013), mass defect filter (Zhu et al., 2007; Zhang et al., 2008b; Ruan and Zhu, 2010; Sleno, 2012; Tian et al., 2015), and neutral loss filer/product ion filter (Schroeder et al., 2004; Zhu et al., 2006, 2011; Hsiao and Urlaub, 2010) based on predicted molecular weights, mass defect values and fragmentation patterns of phase I and phase II metabolites. In addition, untargeted LC-HRMS data processing tools, namely background subtraction (Zhang and Yang, 2008; Zhang et al., 2008a; Zhu et al., 2009; Zhang, 2012; Shekar et al., 2016; Wu et al., 2016) and metabolomics (Giri et al., 2006; Chen et al., 2007; Li et al., 2011; Gonzalez et al., 2015), are often used to search for unpredictable or uncommon metabolites. Once metabolite ions are identified, their MS/MS spectra that are commonly acquired by collision-induced disassociation (CID) and higher-energy collisional dissociation (HCD) can be retrieved from LC-MS/MS datasets.
The last step of metabolite profiling by LC-HRMS is the data interpretation to elucidate metabolite structures based on their mass spectral data and biotransformation knowledge. In the last decade, the sensitivity, accuracy, and scanning speed of LC-HRMS instruments have been greatly improved. Furthermore, metabolite identification software tools make the LC-HRMS data process much faster. As a result, the data acquisition, and data processing in metabolite profiling by LC-HRMS can be accomplished in a relatively short time with superior analytical quality. However, the data interpretation in metabolite identification by LC-HRMS still takes significant time and effort and often fails to provide useful structural information. Indeed, the data interpretation is the bottleneck of drug metabolite profiling and identification by LC-HRMS. For example, if a test drug that has multiple hydroxyls and/or amine groups forms one or more glucuronide metabolites, LC-HRMS analysis most likely cannot determine the metabolic sites of the glucuronidation.
Recently, a ZenoTOF 7600 mass spectrometer equipped with electron-activated dissociation (EAD) is introduced, which is based on the interaction between low-energy electrons and multiply-charged precursor ions. EAD has been applied to the localization and quantification of post-translational modifications of proteins (Bons et al., 2023), top-down characterization of a monoclonal antibody drug (Baba et al., 2021), and identification of modification sites of oxidative metabolites (Che et al., 2023). Results from these studies demonstrate that EAD preserved labile modifications and provided better results in structural characterization.
The primary objective of this study was to apply EAD to identify accurate oxidation and glucuronidation sites of vepdegestrant (ARV-471), a model compound of proteolysis targeting chimeras. In the study, ARV-471 was incubated in dog liver microsomes (DLMs) in the presence of NADPH and uridine-5′-diphosphoglucuronic acid (UDPGA). EAD and CID spectra of vepdegestrant metabolites were acquired by the ZenoTOF 7600 system (EAD-HRMS). Additionally, HCD spectra of these metabolites were recorded using Q Exactive Plus HRMS. As a result, 12 metabolites generated from glucuronidation, oxidation, dealkylation, hydrolysis of ARV-471, and combinations of these biotransformation reactions were detected by LC-HRMS data processing. The interpretation of their EAD spectra allowed us to accurately characterize metabolic modification sites of these phase I and glucuronide metabolites. In contrast, the interpretation of CID and HCD spectra only determined the biotransformation sites of six ARV-471 metabolites. The results demonstrate that the orthogonal fragmentations generated by EAD can reveal labile fragments that are often not seen in CID and HCD spectra, which consequently enables the definitive structural elucidation of some difficult-to-classify metabolites, such as glucuronide conjugates.
Materials and Methods
Chemicals and Reagents
ARV-471 was synthesized in-house, and its identity and purity were assessed using nuclear magnetic resonance spectroscopy, mass spectrometry, and high-performance liquid chromatography (HPLC). DLM was purchased from Corning Gentest (Glendale, AZ). 7-Ethoxycoumarin was purchased from J&K Scientific Ltd. (Shanghai, China). PBS, MgCl2, DMSO, alamethicin, and UDPGA were from Meilunbio (Dalian, China). NADPH was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Ammonium acetate was from Sinopharm Group Co., Ltd. (Shanghai, China). Formic acid was purchased from RHAWN (Shanghai, China). Acetonitrile (ACN) of LC-MS grade was obtained from Sigma-Aldrich (St. Louis, MO). Ultrapure water was freshly prepared with a Millipore purification system (Molsheim, France).
DLM Incubation
ARV-471 and alamethicin were dissolved in DMSO as the 10 mM and 10 mg/ml stock solution, respectively. The stock solution was diluted with ACN to 2 mM and 5 mg/ml working solution. The DLM working solution was prepared by diluting the liver microsome (20 mg/ml, 44.4 μl) with PBS buffer (100 mM, 748.1 μl), and MgCl2 solution (300 mM, 7.5 μl). All incubation contained 180 μl of liver microsome working solution, 1 μl of ARV-471 working solution, and 1 μl of alamethicin working solution. The incubation mixtures were preincubated at 37°C in a thermostatic oscillator for 5 minutes and then 20 μl of the mixture solution of 20 mM NADPH and 20 mM UDPGA were added to initiate the reaction. The final concentration of ARV-471 and DLM was 10 µm and 1 mg/ml. All the samples were duplicates. After 60-minute incubation, 200 μl of the ice-cold ACN was added to terminate the incubation. The tube would be vortexed, the duplicate samples were combined. For the T0 sample: 200 μl of terminating solution (ice-cold ACN) was mixed with 180 μl of liver microsome working solution at first, then 20 μl of the mixture solution of 20 mM NADPH and 20 mM UDPGA, as well as 1 μl of ARV-471 working solution and 1 μl of alamethicin working solution, were added. 7-Ethoxycoumarin, as the positive control (10 μm), was incubated with liver microsomes the same as the test article. The combined sample was centrifuged (15,000 × g, 10 minutes, 4°C) the supernatant was concentrated under an N2 stream at room temperature, and the residues were reconstituted with an appropriate volume of acetonitrile/water (20:80, v/v). The reconstituted solution was transferred into an HPLC vial after centrifugation (15,000 × g, 10 minutes, 4°C).
Sample Analysis by LC-ZenoTOF 7600 Mass Spectrometer
LC separation was performed on a Waters ACQUITY UPLC HSS T3 column (2.1 × 100 mm, 1.8 μm) at a column temperature of 25°C and the flow rate was 0.3 ml/min. Mobile phase A was 0.1% (v/v) formic acid in water and mobile phase B was 0.1% (v/v) formic acid in acetonitrile. An injection of 5 μl reconstituted solution was subjected for analysis. The gradient elution procedure was as follows: 0 minute, 5% B; 2 minutes, 5% B; 10 minutes, 40% B; 11 minutes, 80% B; 16 minutes, 98% B; 18 minutes, 98% B; 18.1 minutes, 5% B; and 20 minutes, 5% B. The samples were analyzed using an information-dependent acquisition method in positive mode with Zeno CID DDA and Zeno EAD DDA on the ZenoTOF 7600 system. The following ion source parameters were applied: a spray voltage of 5.5 kV, a source temperature of 550°C, ion source gas of 1 55 psi, ion source gas of 2 55 psi, curtain gas of 35 psi, and CAD gas of 7 psi. Each MS cycle starts with a survey TOF MS scan in 250 ms from 200 to 1000 Da using the declustering potential at 80 V, followed by a TOF MSMS full scan ranging from 50 to 1000 Da to ensure all fragments were captured for identification. The CID DDA accumulation time was 30 ms, using collision energy at 50 V and collision energy spread at 20 V. The EAD DDA accumulation time was 35 ms, using electron beam current at 8000 nA and electron kinetic energy at 12 eV. Data were acquired with the Zeno trap on and the Zeno threshold was set as 5,000,000.
Sample Analysis by LC-Q Exactive Plus Mass Spectrometer
An LC coupled with Q Exactive Plus HRMS was used to obtain higher-energy collisional dissociation (HCD) spectral data of ARV-471 metabolites. Chromatographic separation was achieved on an Agilent Extend C-18 column (5 µm, 150 mm × 4.6 mm) with a Phenomenex C-18 guard column (5 µm, 10 mm × 4.6 mm). The column temperature was 25°C, and the flow rate was 0.60 ml/min. The mobile phase consisted of a 5 mM aqueous ammonium acetate solution containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The gradient elution procedure was as follows: 0 minutes, 5% B; 2 minutes, 5% B; 10 minutes, 40% B; 11 minutes, 80% B; 16 minutes, 98% B; 18 minutes, 98% B; 18.1 minutes, 5% B; and 20 minutes, 5% B. The mass spectrometer was operated in the positive electrospray ionization (ESI) mode with a mass-to-charge ratio (m/z) of 80–1200 Da scanning range for detection. The optimized MS parameters were as follows: sheath gas, 45 L/min; auxiliary gas, 10 L/min; normalized collision energy, 15, 30, and 45; capillary temperature, 320°C; auxiliary gas heater temperature, 400°C; and capillary voltage, 3.5 kV. LC-HRMS data sets were collected using Xcalibur software (Thermo).
LC-HRMS Data Processing
Acquired LC-HRMS data sets by ZenoTOF 7600 were processed using the Explorer module in SCIEX OS software (version 3.1) and SCIEX Molecule Profiler software (beta version 1.3) for the detection and identification of ARV-471 metabolites. The Molecule Profiler software can help in detecting potential metabolites by annotating ion chromatographic profiles and MS/MS spectral data. A set of biotransformation reactions for processing the data and assigning appropriate metabolites was created based on the structure of ARV-471. Ion chromatographic peaks were considered for the metabolite confirmation and MS/MS fragment interpretation if they corresponded to a predicted mass, or showed at least one characteristic product ion/neutral loss. The following parameters were used to find metabolite ion peaks in the LC-HRMS data. The minimum peak width was 2.5 seconds, the minimum peak intensity was 10,000 cps for TOF MS, the MS m/z tolerance was 10 ppm, and the minimum MS peak intensity was 200 cps.
Acquired LC-HRMS data sets by Q Exactive Plus HRMS were processed using Compound Discoverer software (Thermo) to receive HCD product ion spectral data of ARV-471 metabolites that were detected and characterized using LC- ZenoTOF 7600.
Results
Detection of ARV-471 Metabolites in DLM Incubation
As displayed in Fig. 1, a total of 12 metabolites (M1–M11) of ARV-471 were detected in DLM incubation by LC-ZenoTOF 7600. Retention times, accurate molecular ions, relative abundances, and extracted ion chromatogram of the ARV-471 metabolites are summarized in Supplemental Fig. 1 and Table 1. Based on the LC-HRMS profiling data, approximately 75% of ARV-471 remained after 1 hour of incubation, indicating the metabolism rate of ARV-471 in DLMs was moderate. M4 accounted for 16.64% of the total drug-related components by LC-HRMS signal, and others were relatively minor metabolites. The same metabolites were also detected in DLMs using LC- Q Exactive Plus HRMS.

A combined extracted ion chromatograms of ARV-471 and its metabolites in DLMs incubation sample. The inset is a zoomed extract chromatogram of all minor metabolites (without the parent drug and the major metabolite M4).
Information of ARV-471 and its metabolites detected in DLMs incubation sample for 1 h by LC-HRMS
EAD, CID, and HCD Spectra of ARV-471
The EAD and CID spectra of ARV-471 (C45H49N5O4, 11.64 minutes) exhibited the [M+H]+ ion at m/z 724.3904 (Fig. 2A), and the HCD spectrum exhibited the [M+H]+ ion at m/z 724.3875 (Fig. 2B). Multiple product ions such as m/z 174, 223, 396, and 502 were generated by EAD, CID, and HCD (Fig. 2C). The HCD spectrum of the ARV-471 was like the CID spectrum except for the additional product ion at m/z 145.0654 (Fig. 2C). The EAD spectrum of ARV-471 displayed several unique, minor product ions, such as the ions at m/z 341.1645, 424.2343, 451.2808, and 612.3534, which provided key information for determining oxidation and glucuronidation sites of ARV-471 metabolites (Figs. 3–6 and Supplemental Figs. 2–12).

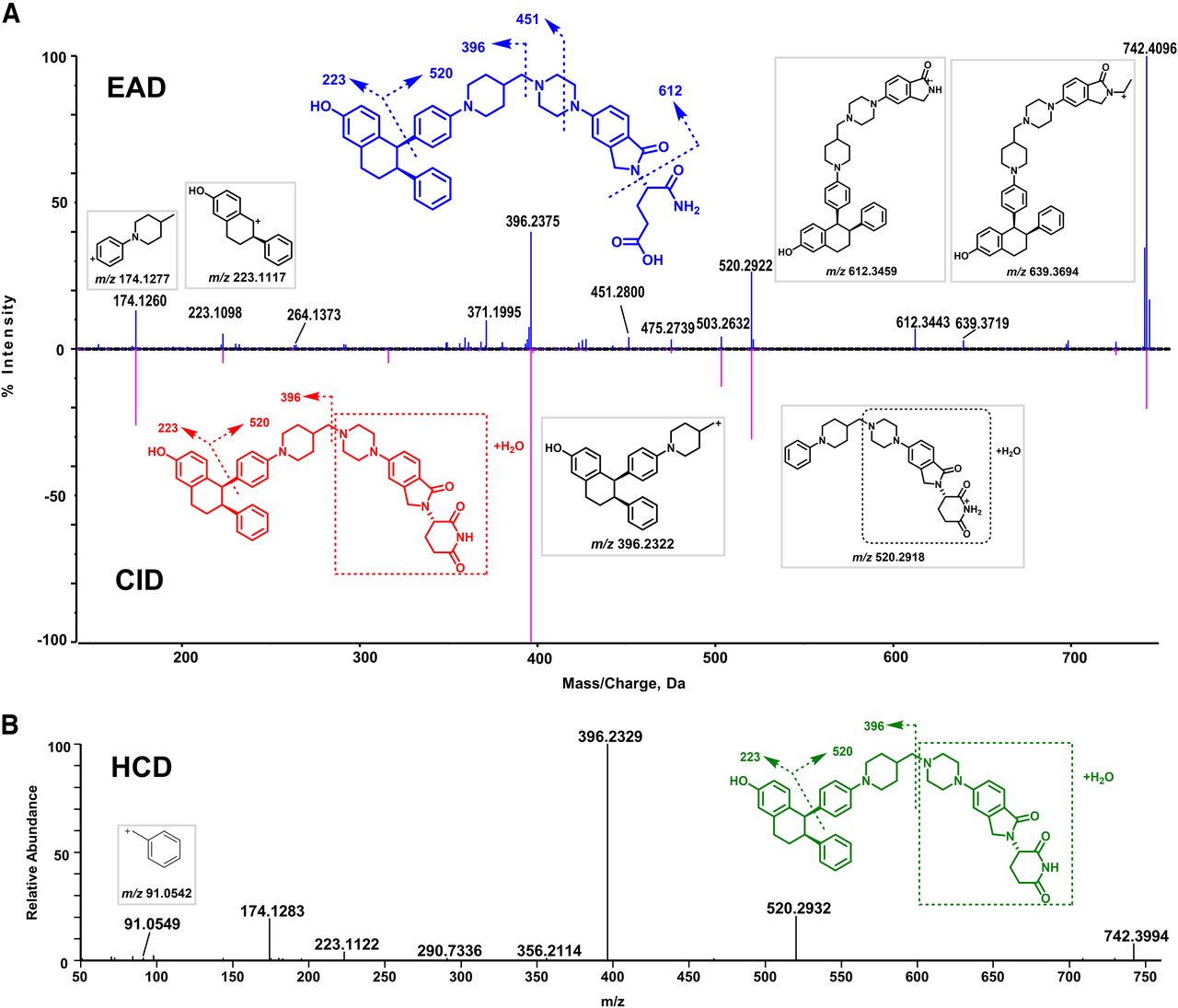
EAD, CID, and HCD spectra and structure of the parent drug ARV-471. (A) EAD (top) and CID (bottom) spectra of ARV-471. (B) HCD spectrum of ARV-471. (C) The proposed structure and fragmentation interpretation of ARV-471. The fragment ions produced by EAD, CID, and HCD are presented in red, those produced by EAD only are presented in blue, and those produced by HCD only are presented in green.

EAD, CID, and HCD spectra and proposed structures of M5. (A) EAD (top) and CID (bottom) spectra of M5. The structure inferred from the EAD data are displayed in blue. There are possibly two metabolite structures from the hydrolysis of individual amide bonds, and one possible structure is displayed here. The proposed structure of M5 based on CID spectral interpretation is indicated in red. (B) HCD spectrum and proposed structure of M5 are displayed in green.

EAD, CID, and HCD spectra and structure of M7 (A) and M2-1 (B). The structures inferred from the EAD data are displayed in blue, the structure identified by CID is indicated in red, and the structure identified by HCD is indicated in green.

EAD, CID, and HCD spectra and structure of M3. (A) EAD (top) and CID (bottom) spectra of M3. The structure inferred from the EAD data are displayed in blue and the structure identified by CID is indicated in red. (B) HCD spectrum of M3 with the proposed structure in green.

EAD, CID, and HCD spectra and structure of M10 (A) and M4 (B). The structures inferred from the EAD data are displayed in blue and the structures identified by CID are indicated in red, and the structures identified by HCD are indicated in green.
Glutarimide Hydrolysis of ARV-471 to M5 and Its Further Metabolism
Four metabolites (M5, M7, M2-1, and M2-2) were formed via glutarimide hydrolysis (M5) and further methylation (M7) and glucuronidation (M2-1 and M2-2) of M5. Their EAD and CID spectra, fragment assessments, and proposed structures are displayed in Figs. 3 and 4 and Supplemental Fig. 2.
M5
The EAD, CID, and HCD spectra of M5 are shown in Fig. 3. M5 (C45H51N5O5, 11.55 minutes) displayed the [M+H]+ ion at m/z 742.4096, which was 18.0192 Da (+H2O) greater than that of ARV-471 (Fig. 3A), suggesting it was a hydrolysis product. However, its metabolism site cannot be determined based on the CID or HCD spectral data. The CID spectrum of M5 showed the ions m/z 396.2375, 223.1098, and 174.1260, which were also the product ions of ARV-471 (Fig. 2A), indicating that the reaction took place in the right part of the box out (Fig. 3A). However, the fragments displayed in the CID spectrum did not allow to give accurate identification of the hydrolysis site of ARV-471. The HCD spectrum displayed the same fragmentations as CID (Fig. 3B). In contrast, the EAD spectrum of M5 indicated an additional minor product ion at m/z 612.3443, which allowed us to determine that the metabolic site was on the glutarimide ring of M5 (Fig. 3A).
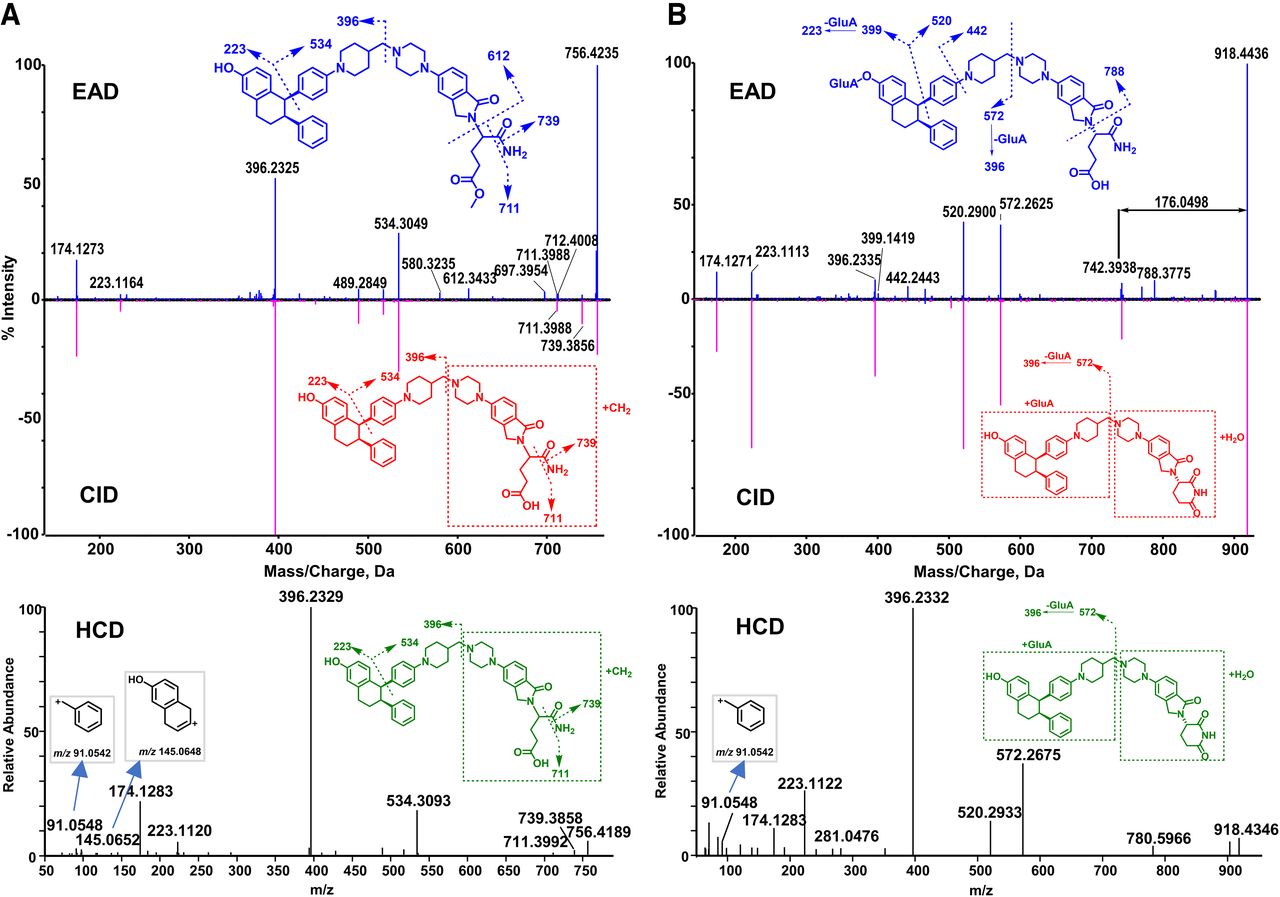
M7
M7 (C46H53N5O5, 11.65 minute) displayed the precursor ion at m/z 756.4235 (Fig. 4A), which was 14.0139 Da (+CH2) greater than the molecular ion of M5 (m/z 742.4096) (Fig. 3A), indicating that it was a methylation product of M5. The same major product ions at m/z 174, 223, 396, 534, 711, and 739 were displayed in EAD, CID, and HCD spectra. The formation of the product ion at m/z 396 that was also generated from M5 indicates that the hydrolysis and methylation occurred on the right side of the molecule. Furthermore, the unique product ion at m/z 612.3433 displayed in the EAD spectrum of M7 indicates that the hydrolysis and methylation sides are located on the glutarimide ring. Based on the EAD spectral interpretation, we propose that M7 was a product of carboxylic acid methylation. However, there is no experimental data to support that the methylation was an enzymatic reaction although methylation metabolites of carboxylic acid have been reported (Liang et al., 2009; Zheng et al., 2021). In contrast, the CID or HCD spectrum of M7 did not exhibit the informative product ion at m/z 612.3433, consequently, it failed to provide the defined structural information as the EAD spectrum (Fig. 4A).
M2-1 and M2-2
The EAD, CID, and HCD spectra of M2-1 (C51H59N5O11, 9.95 minutes) and M2-2 (C51H59N5O11, 10.12 minutes) are depicted in Fig. 4B and Supplemental Fig. 2, respectively. M2-1 displayed the [M+H]+ ion at m/z 918.4436, 176.0498 Da (+C6H8O6) greater than the molecular ion of M5 (Fig. 4B), indicating that it is a glucuronide metabolite of M5. A few major product ions of M2-1 (such as m/z 174, 223, 396, 520, and 572) are displayed in its EAD, CID, and HCD spectra (Fig. 4B). The product ion at m/z 572.2625 was 176.0290 Da (+C6H8O6) greater than m/z 396.2335 that is a major fragment ion of the parent, indicating that the glucuronide acid binding site was in the left part and the hydrolysis occurred in the right part of M2-1. Additionally, the EAD spectrum of M2-1 displayed a few unique product ions at m/z 788.3775, 442.2443, and 399.1419. The ion at m/z 788.3775 was 176.0342 Da (+C6H8O6) greater than the M5 product ion at m/z 612.3433, revealing that the hydrolysis took place on the glutarimide ring (Fig. 4B). The diagnostic ion at m/z 399.1419 only shown in the EAD spectrum confirmed that the glucuronide conjugation occurred on the phenol hydroxyl group. Due to the lack of the informative product ions at m/z 788 and 399 in the CID and HCD spectra of M2-1, the interpretation of the CID and HCD product ion spectra failed to provide the metabolism site information of M2-1 (Fig. 4B).
N-Dealkylation of ARV-471 to M8 and M11 and Their Further Metabolism
ARV-471 underwent a dealkylation reaction to form M8 (C28H31NO2, 11.69 minutes) and M11 (C17H20N4O3, 4.91 minutes), which further underwent phase I and II metabolism to form M1, M3, and M10. Their EAD, CID, and HCD spectra, fragment assignments, and proposed structures are displayed in Figs. 5 and 6A, and Supplemental Figs. 3–8.
M8 and M11
Based on protonated molecular ions of M8 and M11 (Supplemental Figs. 3–6; Table 1) and biotransformation knowledge, M8 and M11 were readily determined as two metabolites from N-dealkylation of ARV-471.
M1
M1 (C19H22N4O4, 7.74 minutes) showed the [M+H]+ ion at m/z 371.1383 (Supplemental Fig. 7), which was 41.9708 Da (+CH2CO) greater than the [M+H]+ ion of M11 (m/z 329.1675), suggesting that M1 is the N-acetylation product of M11. The EAD, CID, and HCD spectra of M1 showed the product ions at m/z 218, 260, and 298 (Supplemental Figs. 7 and 8). The product ion at m/z 260.1393 of M1 was 42.0118 Da greater than m/z 218.1275, a product ion of M11, indicating the acetylation site on the piperazine ring.
M3
M3 (C34H39NO8, 10.09 minutes) was observed with [M+H]+ at m/z 590.2871 (Fig. 5), which was 176.0372 Da (+C6H8O6) greater than the molecular ion of M8 (m/z 414.2499) (Supplemental Fig. 3), indicating it may be a glucuronide metabolite of M8. There are three potential glucuronidation sites (i.e., the phenol hydroxyl, amino, and alethic hydroxyl groups) in M3. However, major product ions m/z 192, 223, and 414 shown in EAD, CID, and HCD spectral data were not able to locate the glucuronidation site. The EAD spectrum of M3 revealed several unique minor product ions, such as m/z 518.2204, 504.2000, 327.1448, and 116.1039 (Fig. 5A). The product ion m/z 327.1448 was crucial in revealing the glucuronide conjugation in the phenol hydroxyl group. The product ion at m/z 116.1039 was also observed in the EAD spectrum of M8 (Supplemental Fig. 3) but not in those of other metabolites, indicating that both M3 and M8 had the same functional group.
M10
M10 (C28H29NO3, 12.081 minutes) displayed precursor ion m/z 428.2241 (Fig. 6A), which was 13.9742 Da (+O, –2H) greater than M8 (m/z 414.2499) (Supplemental Fig. 3), indicating M10 was a metabolite from oxidation and dehydrogenation of M8. The principal product ions (such as m/z 206, 223, and 410) of M10 were displayed in EAD, CID, and HCD spectra (Fig. 6A). The product ion at m/z 206.1186 was 13.9787 Da (+O–2H) greater than the product ion (m/z 192.1399) of M8 (Supplemental Fig. 3), suggesting the oxidation and reduction occurred on multiple function groups (Fig. 6A). In contrast to the CID and HCD spectra, the EAD spectrum of M10 displayed an extra minor product ion at m/z 112.0765, which was 4.0315 Da (+O–2H–H2O) less than the product ion at m/z 116.1080 of M8 (Supplemental Fig. 3). Because M10 was eluted much later than M8 under an acidic mobile phase system (Fig. 1), we proposed M10 to be a piperidine lactam metabolite via lactam formation with alkalinity reduction (Zhu et al., 2016).
Glucuronidation of ARV-471 to M4
The EAD, CID, and HCD spectra of M4 are displayed in Fig. 6B. M4 (C51H57N5O10, 10.49 minutes) displayed the precursor ion at m/z 900.4183, which was 176.0270 Da (+C6H8O6) greater than ARV-471 (m/z 724.3913), indicating that it is a glucuronide metabolite of ARV-471. Multiple product ions (such as m/z 572, 502, 399, 396, 223, and 174) were displayed in EAD, CID, and HCD spectral data (Fig. 6B). The ion at m/z 572.2689 was 176.0341 Da (+C6H8O6) more than m/z 396.2348, the ion at m/z 399.1461 was 176.0297 Da (+C6H8O6) more than m/z 223.1164, and other ions were the same as those observed in ARV-471, suggesting the glucuronide acid binding on the phenol hydroxyl group. In addition, the EAD spectrum of M4 displayed unique product ions, such as the ions at m/z 788.3861 and 424.2325. The product ion at m/z 788.3861 was 176.0327 Da (+C6H8O6) greater than the ion at m/z 612.3534 of ARV-471 and the ion at m/z 424.2325 was the same as that of ARV-471, which helped to identify the glucuronide conjugation on the phenol hydroxyl group (Fig. 6B).
Metabolites Formed via ARV-471 Oxidation
Two metabolites (M6 and M9) were detected as products of ARV-471 oxidation. Their EAD, CID, and HCD spectra and proposed structures based on EAD, CID, and HCD fragmentation assessments are displayed in Supplemental Figs. 9–12.
M6
M6 (C45H49N5O5, 11.590 minutes) displayed [M+H]+ m/z 740.3787 (Supplemental Fig. 9), which was 15.9883 Da (+O) larger than m/z 724.3904 of ARV-471 (Fig. 2A), suggesting it is an oxidation product of ARV-471. Both Zeno CID DDA and Zeno EAD DDA data provided a few principal product ions (such as m/z 722.3692, 518.2730, 500.2651, 412.2277, 394.2171, 223.1116, and 172.1113) that matched identically (Supplemental Figs. 9 and 10). Among them, m/z 722.3692 was the neutral loss of H2O (18.0095 Da) from m/z 740.3787; m/z 518.2730 was 15.9818 Da (+O) larger than m/z 502.2912 of ARV-471; m/z 500.2651 was 18.0079 Da (–H2O) smaller than m/z 518.2730; m/z 412.2277 was 15.9866 Da (+O) more than m/z 396.2411 of ARV-471; m/z 394.2171 was 18.0106 Da (–H2O) less than m/z 412.2277; m/z 223.1116 was observed in ARV-471, and m/z 172.1113 was 2.0237 Da (+O–H2O) less than m/z 174.1350 of ARV-471, providing sufficient details to the oxidation site on the piperidine ring of M6.
M9
M9 (C45H47N5O4, 11.722 minutes) displayed [M+H]+ m/z 722.3801 (Supplemental Fig. 11), which was 2.0103 Da (–2H) less than m/z 724.3904 of ARV-471, implying it is an oxidation product of ARV-471. EAD, CID, and HCD data provided the major product ions (such as m/z 500, 394, 223, and 172) that matched identically (Supplemental Figs. 11 and 12). Among these ions, m/z 500, 394, and 172 were 2 Da (–2H) less than m/z 502, 396, and 174 of ARV-471; m/z 223.1122 was also observed in ARV-471, providing adequate details of the oxidation site on the piperidine ring of M9.
Discussion
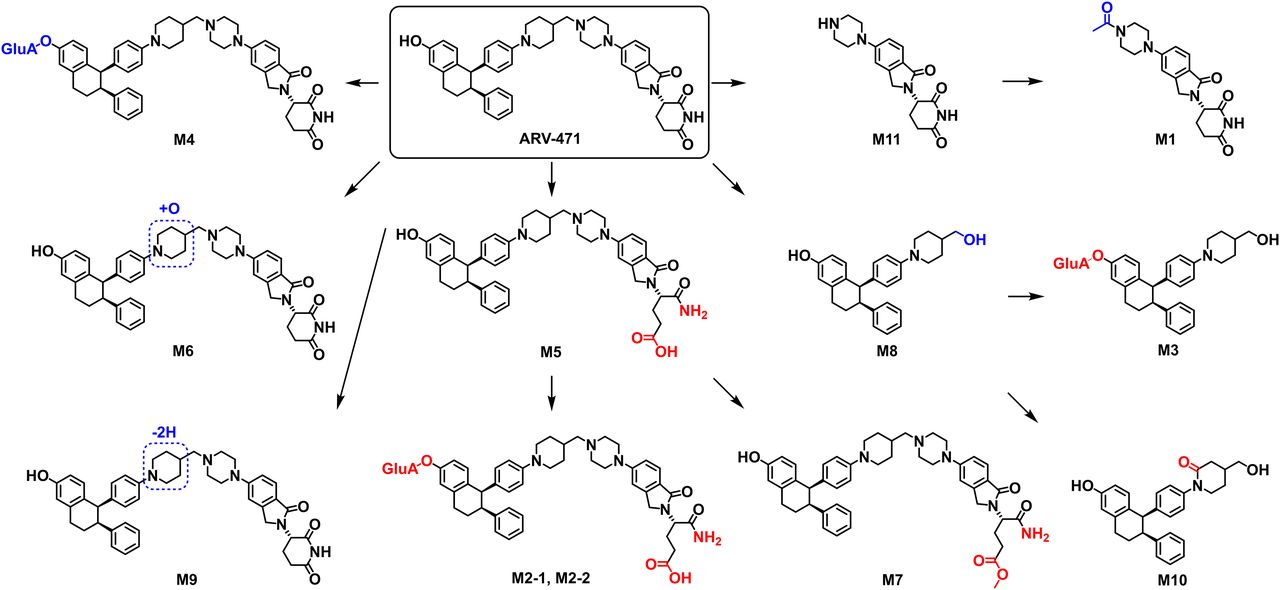
In this study, ARV-471 was used as a model compound to evaluate the effectiveness of EAD in the structural characterization of glucuronides and phase I metabolites. ARV-471 was incubated in DLMs followed by sample analysis using LC-ZenoTOF 7600 mass spectrometer. As a result, accurate EAD spectral data of ARV-471 metabolites were generated. For a comparison purpose, CID and HCD spectra of the same metabolites were also generated (Figs. 3–6 and Supplemental Figs. 2–12). A total of 12 metabolites of ARV-471 were detected in the DLMs (Fig. 1). Based on the interpretation of the EAD spectra, biotransformation pathways and specific metabolic modification sites of the ARV-471 were identified (Fig. 7; Table 1), including piperazine N-dealkylation (M11, M8, M1, M3, and M10), glutarimide hydrolysis (M5, M7, M2-1, and M2-2), phenol glucuronidation (M4, M2-1, M2-2, and M3), piperidine oxidation (M6 and M9), and piperidine lactam formation (M10). Based on the relative peak intensities (Fig. 1) and metabolite structures, it was determined that the phenol glucuronidation to form M4 was the single major metabolic pathway of ARV-471 in DLMs (Figs. 1 and 7). In addition, ARV-471 underwent multiple minor metabolic pathways, including piperazine N-dealkylation, piperidine oxidation, and glutarimide hydrolysis. The interpretation of the CID or HCD spectra failed to identify the modification sites of the O-glucuronidation (M2-1, M2-2, and M3), glutarimide hydrolysis (M5, M2-1, M2-2, and M7), and piperidine lactam formation (M10) of ARV-471 (Fig. 7).

Proposed metabolic pathway of ARV-471 in DLMs incubation sample. The structures identified from the EAD data are displayed in red.
In general, glucuronidation conjugation sites are difficult to characterize by LC-HRMS because glucuronide conjugates readily generate the major product ion of unconjugated drugs via a neutral loss of 176.032 (the glucuronic moiety). For example, M3 has three functional groups (phenol, aliphatic hydroxyl, and tertiary amine) that are potentially glucuronidation sites (Fig. 5). The CID spectrum of M3 displayed a major product ion at m/z 414.2410 via a neutral loss of 176.0461 and a few sequential product ions of the ion at m/z 414.2410, which were not useful for identifying the glucuronidation site of M3. The HCD spectrum of M3 showed similar product ions to those of CID except that HCH generated smaller product ions. In contrast, the EAD spectrum of M3 revealed the ion at m/z 327.1448, which indicated that the glucuronidation site of M3 is the phenyl group (Fig. 5). Similarly, EAD spectra of other ARV-471 glucuronides (M2-1 and M2-2) provided the key informative fragments (m/z 399.1419) that allowed the identification of the phenyl group of ARV-471 as their specific glucuronidation site (Fig. 4B and Supplemental Fig. 2).
In addition to facilitating the elucidation of the glucuronidation sites, EAD generated informative fragments that were crucial for determining modification sites of phase I metabolism of ARV-471 such as glutarimide hydrolysis (M5, M7) (Figs. 3 and 4A) and piperidine lactam formation (M10) (Fig. 6A). Those informative fragment ions were not shown in the CID and HCD spectra. For example, the EAD spectrum of M5, the hydrolysis metabolite, showed a product ion at m/z 612.3443 (Fig. 3A), which was also displayed in the EAD spectrum of ARV-471 (Fig. 2A), indicating the hydrolysis site was in the glutarimide function group. However, the glutarimide hydrolysis site of M5 was not determined via the CID and HCD spectral interpretation due to the lack of the product ion at m/z 612.3443. Comparisons of EAD, CID, and HCD spectra of ARV-471 metabolites (Fig. 2A) indicated that EAD involved the interaction of an electron beam with a molecular ion and underwent orthogonal fragmentation, resulting in the cleavage of more bonds and the formation of smaller ions than CID and HCD.
As shown in the EAD spectral data of ARV-471 and its metabolites (Figs. 2–6), EAD generated several unique product ions that were not shown in the CID and HCD spectra, one or two of which provided crucial information for the definitive identification of the functional groups that were metabolically modified. In general, HCD and CID spectra of ARA-471 metabolites are similar. The major difference between HCD and CID is that HCD can generate much smaller fragments than CID, which did not provide valuable information for accurately identifying metabolic sites of ARV-471.
In summary, this study aimed to evaluate new EAD technology in the determination of metabolic modification sites of small molecule drugs by using ARV-471 as a model compound that consists of multiple functional groups such as piperidine, benzene, phenol, piperazine, and glutarimide. Based on EAD spectral interpretation, modification sites and formation pathways of 12 ARV-471 metabolites were identified, including phenyl glucuronidation, piperidine mono-oxidation, piperidine dehydrogenation, glutarimide hydrolysis, piperazine N-dealkylation, and piperidine lactam formation. In contrast, CID and HCD spectral interpretation only identified the accurate metabolism sites of six metabolites (M8, M11, M1, M6, M9, and M4). M8, M11, and M1 were generated via piperazine N-dealkylation, which was readily identified based on their molecule weights. The results demonstrate that the orthogonal fragmentations generated by EAD can reveal labile fragments that are often not seen in CID and HCD spectra, which consequently enabled the definitive structural elucidation of some difficult-to-classify metabolites. However, because this study only dealt with a few types of metabolic modifications of a single test compound, the advantages, and limitations of EAD over CID and HCD remain to be further explored via the identification of metabolites that have various structures and formed via all types of biotransformation reactions.
Acknowledgments
The authors thank Sciex for the assistance with the ZenoTOF 7600 system.
Data Availability
The authors declare that all the data supporting the findings of this study are available within the paper.
Authorship Contributions
Participated in research design: He, Zheng, Tang, Diao, Zhu.
Conducted experiments: He, Hou.
Contributed to method development: Hou, Long, Jones.
Performed data analysis: He, Hou, Long, Zheng, Tang, Diao, Zhu.
Wrote or contributed to the writing of the manuscript: He, Hou, Long, Zheng, Tang, Diao, Zhu.
Footnotes
- Received January 17, 2024.
- Accepted March 28, 2024.
The study was partly funded by a grant from the National Natural Science Foundation of China [Grant 82373938].
P.H., Z.L., and E.J. are the employees of Sciex, C.T. is the employee of XenoFinder Co., Ltd. M.Z. is the employee of MassDefect Technologies. The other authors declare no conflicts of interest.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- ARV-471
- vepdegestrant
- CID
- collision-induced dissociation
- DDA
- data-dependent acquisition
- DLM
- dog liver microsome
- EAD
- electron-activated dissociation
- HCD
- higher-energy collisional dissociation
- IDA
- information-dependent acquisition
- LC-HRMS
- liquid chromatography-high resolution mass spectrometry
- LC-MS
- liquid chromatography-mass spectrometry
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- m/z
- mass-to-charge ratio
- UDPGA
- uridine-5′-diphosphoglucuronic acid
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}