


Abstract
Gemfibrozil, a fibrate hypolipidemic agent, is eliminated in humans by glucuronidation. A gemfibrozil glucuronide has been reported to show time-dependent inhibition of cytochrome P450 2C8. Comprehensive assessment of the drug interaction between gemfibrozil and cytochrome P450 2C8 substrates requires a clear understanding of gemfibrozil glucuronidation. However, the primary UDP-glucuronosyltransferase (UGT) isozymes responsible for gemfibrozil glucuronidation remain to be determined. Here, we identified the main UGT isozymes involved in gemfibrozil glucuronidation. Evaluation of 12 recombinant human UGT isozymes shows gemfibrozil glucuronidation activity in UGT1A1, UGT1A3, UGT1A9, UGT2B4, UGT2B7, and UGT2B17, with UGT2B7 showing the highest activity. The kinetics of gemfibrozil glucuronidation in pooled human liver microsomes (HLMs) follows Michaelis-Menten kinetics with high and low affinity components. The high affinity Km value was 2.5 μM, which is similar to the Km value of gemfibrozil glucuronidation in recombinant UGT2B7 (2.2 μM). In 16 HLMs, a significant correlation was observed between gemfibrozil glucuronidation and both morphine 3-OH glucuronidation (r = 0.966, p < 0.0001) and flurbiprofen glucuronidation (r = 0.937, p < 0.0001), two reactions mainly catalyzed by UGT2B7, whereas no significant correlation was observed between gemfibrozil glucuronidation and either estradiol 3β-glucuronidation and propofol glucuronidation, two reactions catalyzed by UGT1A1 and UGT1A9, respectively. Flurbiprofen and mefenamic acid inhibited gemfibrozil glucuronidation in HLMs with similar IC50 values to those reported in recombinant UGT2B7. These results suggest that UGT2B7 is the main isozyme responsible for gemfibrozil glucuronidation in humans.
Gemfibrozil, 5-(2,5-dimethylphenoxy)-2,2-dimethylpentanoic acid (Fig. 1), is a fibric acid derivative widely used as a lipid-regulating agent. Gemfibrozil undergoes extensive glucuronidation and oxidative metabolism (Nakagawa et al., 1991), with gemfibrozil 1-O-β-glucuronide as one of the major metabolites in humans. Studies show that 32% of a gemfibrozil dose is excreted as a gemfibrozil conjugate in urine by 24 h after administration (Nakagawa et al., 1991). A recent report showed that the gemfibrozil glucuronide time-dependently inhibits CYP2C8 in vitro (Ogilvie et al., 2006). In a clinical study, gemfibrozil increased the plasma concentration of pioglitazone (Deng et al., 2005), repaglinide (Niemi et al., 2003), and cerivastatin (Backman et al., 2002), which are primarily metabolized by CYP2C8 and CYP3A4. In addition, gemfibrozil and its glucuronide are indicated to inhibit organic anion transporting polypeptide 1B1 (Shitara et al., 2004). Comprehensive assessment of the drug interaction between gemfibrozil and substrates of CYP2C8 and organic anion transporting polypeptide 1B1 thus requires a clear understanding of the formation of gemfibrozil glucuronide. Although UDP-glucuronosyltransferase (UGT) isozymes such as UGT1A1, UGT1A3, UGT1A9, UGT2B7, and UGT2B15 possess catalytic activity for gemfibrozil glucuronidation, the major UGT isozyme responsible for this glucuronidation remains to be determined (Prueksaritanont et al., 2002a). Examination of the main UGT isozymes involved in the formation of the acyl glucuronide of gemfibrozil, which was shown to covalently bind to protein (Sallustio et al., 1997), is required to evaluate possible factors having an impact on acyl glucuronide-associated toxicity. Furthermore, identification of the main isozymes involved in drug metabolism is crucial to understanding the variations in drug concentration that lead to altered drug efficacy and/or toxicity. Therefore, we were interested in identifying the UGT isozyme responsible for gemfibrozil glucuronidation.
Identifying the main UGT isozymes involved in the glucuronidation of drugs is hampered by the paucity of information on specific inhibitors for the respective UGT isozymes. However, Uchaipichat et al. have reported that diclofenac is a relatively potent UGT2B7 inhibitor (Uchaipichat et al., 2004) and fluconazole is a selective inhibitor of UGT2B7 (Uchaipichat et al., 2006). The glucuronidation of β-estradiol at the 3-OH position was reported to be a probe reaction for UGT1A1, whereas that of trifluoperazine, propofol, and morphine at the 3- and 6-OH positions was reported to be probe reactions for UGT1A4, UGT1A9, and UGT2B7, respectively (Stone et al., 2003; Miners et al., 2006; Mano et al., 2007a). In addition, recent findings suggest that flurbiprofen is catalyzed mainly by UGT2B7 (Mano et al., 2007b) and is also a potent inhibitor of UGT2B7 (Bauman et al., 2005). Mefenamic acid has also been reported as a potent inhibitor of UGT2B7 (Mano et al., 2007a). These reports lead us to use flurbiprofen and mefenamic acid as the inhibitors in our study, although it is not yet known whether these inhibitors are completely selective for UGT2B7. Identification of the UGT isozymes responsible for glucuronidation is assisted by correlation studies between these probe reactions in individual human liver microsomes (HLMs) and gemfibrozil glucuronidation, as well as comparison of the kinetic parameters and inhibition potency of glucuronidation between HLMs and recombinant human UGT isozymes.

Chemical structures of gemfibrozil and its 1-O-β-glucuronide. A, gemfibrozil. B, gemfibrozil 1-O-β-glucuronide.
Here, we determined the UGT isozyme responsible for gemfibrozil glucuronidation in humans. The activities and kinetics for glucuronidation were determined using HLMs and recombinant human UGTs. We also performed an inhibition study using HLMs and a correlation study using the typical substrate for the UGT isozyme.
Materials and Methods
Chemicals and Reagents. Racemic flurbiprofen and mefenamic acid were purchased from Sigma (St. Louis, MO). Gemfibrozil and gemfibrozil 1-O-β-glucuronide were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Pooled (mixed gender, n = 50) and individual HLMs were obtained from Xenotech (Kansas City, KS), and recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 were purchased from BD Biosciences (Bedford, MA). All the other chemicals were of analytical grade.
Kinetic Study in HLMs. Gemfibrozil (0.5-400 μM) was preincubated with pooled HLMs (0.1 mg of protein/ml) at 37°C for 5 min in a final volume of 0.25 ml of Tris-HCl buffer (50 mM, pH 7.5) containing 8 mM MgCl2, and 25 μg/ml alamethicin. The alamethicin concentration used in this study (25 μg/ml) is that recommended by the supplier and is similar to that in a previous report (Kuehl and Murphy, 2003). After preincubation of the reaction mixture, the reaction was started by adding UDP glucuronic acid to the mixture to a final concentration of 5 mM. After incubation at 37°C for 10 min, the reaction was terminated by precipitating the protein by adding 1.0 ml of acetonitrile. An internal standard [0.02 ml of deuterium gemfibrozil glucuronide (1 μg/ml)] was then added, and the samples were centrifuged at 1870g for 10 min to obtain the supernatant, followed by evaporation of the supernatant to dryness using nitrogen gas. Samples were reconstituted in 0.2 ml of 0.1% formic acid/acetonitrile (6:4, v/v). Aliquots (30 μl) were then injected into a high-performance liquid chromatography with tandem mass spectrometry system.
Glucuronidation by Recombinant UGTs. Gemfibrozil glucuronidation was measured in recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17. Substrate concentrations of 1 and 100 μM and respective protein concentrations of 0.1 and 0.05 mg of protein/ml were used. All the isozymes were left to react for 20 min. It should be noted that a simple comparison of the glucuronidation activity among UGT isozymes is not sufficient to elucidate the main UGT involved in the liver because the UGT content in each recombinant UGT isozyme could not be determined, and the relative abundance of UGTs in the human liver has not been determined. A kinetic study for gemfibrozil glucuronidation by recombinant UGT1A3 and UGT2B7 was also conducted by incubating gemfibrozil with UGT1A3 (0.1 mg of protein/ml) or UGT2B7 (0.05 mg of protein/ml) for 15 min. The ranges of substrate concentrations used to obtain kinetic profiles were 5 to 1000 μM and 1to200 μM in recombinant UGT1A3 and UGT2B7, respectively.
Correlation Study in HLMs. The glucuronidation of gemfibrozil was measured in microsomes of 16 individual human livers. The activity of UGT1A1-catalyzed estradiol 3β-glucuronidation and UGT1A9-catalyzed propofol glucuronidation was 226 to 1360 and 123 to 2330 pmol/min/mg protein, respectively. The activity of UGT2B7-catalyzed morphine 3-OH glucuronidation and flurbiprofen glucuronidation [flurbiprofen (R)-glucuronide formation] was 3060 to 9670 and 750 to 2024 pmol/min/mg protein, respectively. The substrate and HLM concentrations were 10 μM and 0.1 mg of protein/ml, respectively. The reaction mixture was incubated for 10 min at 37°C. The correlation between gemfibrozil glucuronidation and estradiol 3β-glucuronidation, propofol glucuronidation, morphine glucuronidation at the 3-OH position, and flurbiprofen glucuronidation was determined using Pearson's moment method with Prism version 3.02 (Graph Pad Software, San Diego, CA). Glucuronidation activities of β-estradiol at the 3-OH position, of propofol, and of morphine at the 3-OH position were provided by the manufacturer as typical reference activities for UGT1A1, UGT1A9, and UGT2B7, respectively. Data for flurbiprofen glucuronidation were based on a previously published study using the same panel of HLMs (Mano et al., 2007b). A p value of less than 0.05 was considered statistically significant.
Chemical Inhibition in HLMs. Gemfibrozil glucuronidation in pooled HLMs was measured in the absence and presence of either mefenamic acid (Mano et al., 2007a) or racemic flurbiprofen (Bauman et al., 2005), two potent inhibitors for UGT2B7. To determine the IC50 values of these inhibitors, gemfibrozil (1 μM) was incubated in the absence and presence of either mefenamic acid (0.1-10 μM) or flurbiprofen (1-100 μM). Incubation was performed for 10 min using a protein concentration of 0.1 mg of protein/ml.
Assay. The peak areas of gemfibrozil glucuronide and its internal standard were analyzed by high-performance liquid chromatography with a tandem mass spectrometry system. A TSQ7000 Quantum Ultra or TSQ7000 triple quadrupole mass spectrometer with an atmosphere pressure ionization source (Thermo Fisher Scientific, Waltham, MA) was used. The atmosphere pressure ionization source was fitted with an electrospray ionization inlet to ionize the analytes. The electron spray voltage was set to 4.5 kV, and the heated capillary temperature was maintained at 300°C. In the negative ion mode, gemfibrozil glucuronide and its internal standard were subjected to selected reaction monitoring, with transmitted molecular ions at m/z 425.0 and 431.1, respectively. These ions were subjected to collision-activated dissociation using argon (1.5 mtorr) at 42 eV, followed by monitoring of the product ions at m/z 121.0 for both gemfibrozil glucuronide and the internal standard. Chromatographic separation was achieved using an XTerra MS C18 column (4.6 × 50 mm, 5 μm) (Waters, Milford, MA), with a mobile phase of 0.1% formic acid/acetonitrile (6:4, v/v) at a flow rate of 0.5 ml/min. The standard curve for the gemfibrozil glucuronide was linear from 0.02 to 10 μM, and the correlation coefficient was >0.99. The accuracy and precision of the back-calculated values for each concentration were less than 15%.
Data Analysis. For determination of kinetic parameters, substrate concentration (S) and velocity (V) data for gemfibrozil glucuronidation in pooled HLMs were fitted to biphasic Michaelis-Menten kinetics (eq. 1), and those for recombinant UGT1A3 and UGT2B7 to monophasic Michaelis-Menten kinetics (eq. 2), to obtain the concentration representing the Michaelis constant (Km) and the maximum velocity (Vmax). Prism version 3.02 software (Graph Pad Software) was used for the calculations. The intrinsic clearance value in pooled HLMs was calculated from Vmax.1/Km.1 + Vmax.2/Km.2. 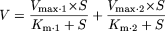
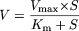
Results
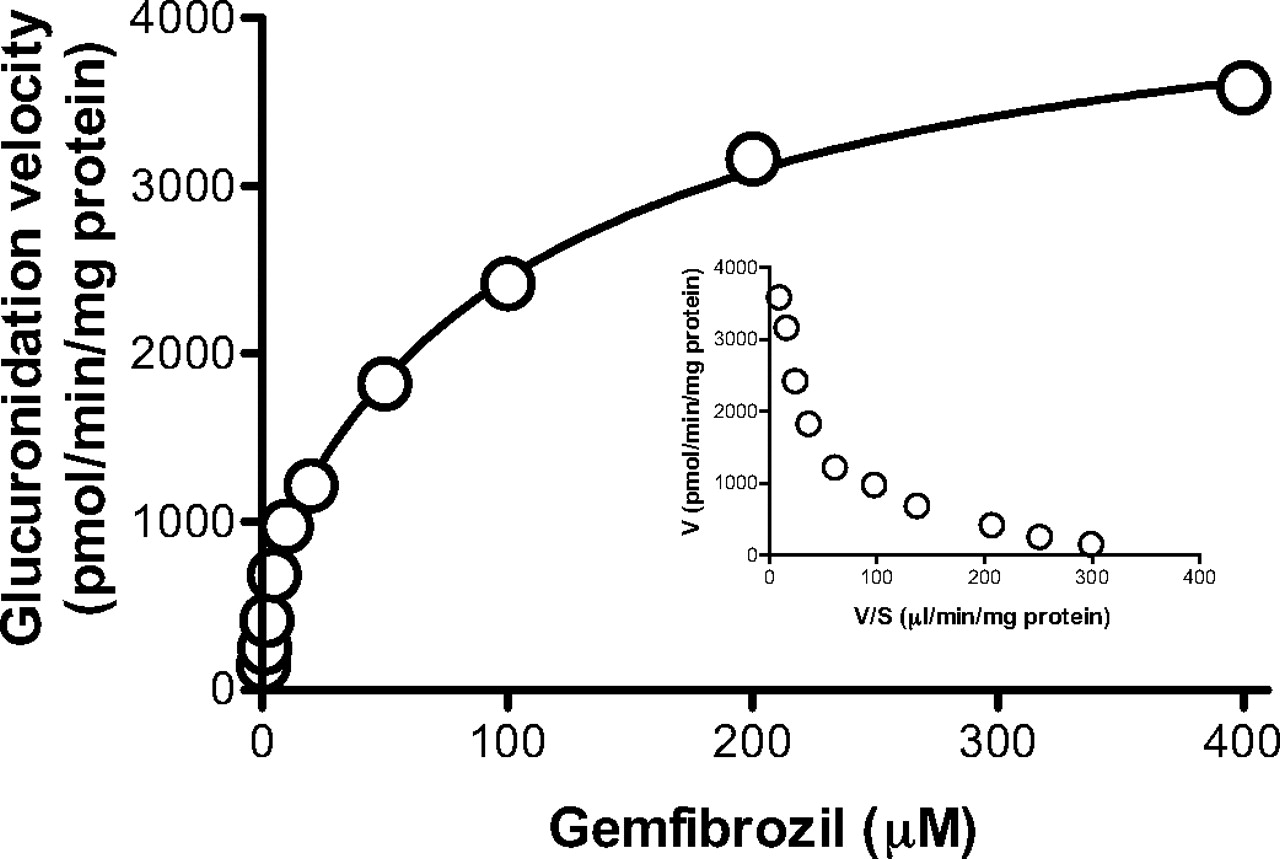
Kinetic Study in HLMs. We first investigated the kinetics of gemfibrozil glucuronidation in pooled HLMs. The substrate concentration-glucuronidation velocity curve followed typical Michaelis-Menten kinetics with high and low affinity components (see the Eadie-Hofstee plot as an inset in Fig. 2). The Km values for high and low affinity components were 2.5 ± 0.7 and 121 ± 16 μM, respectively. The respective Vmax values were 812 ± 106 and 3670 ± 101 pmol/min/mg protein (mean ± computer-calculated S.E.). The intrinsic clearance value was 355 μl/min/mg protein.

Kinetics of gemfibrozil glucuronidation in HLMs. Gemfibrozil (0.5-400 μM) was incubated with pooled HLMs (0.1 mg of protein/ml) for 10 min. The Eadie-Hofstee plot is shown as an inset. Data represent the mean of duplicate determination.
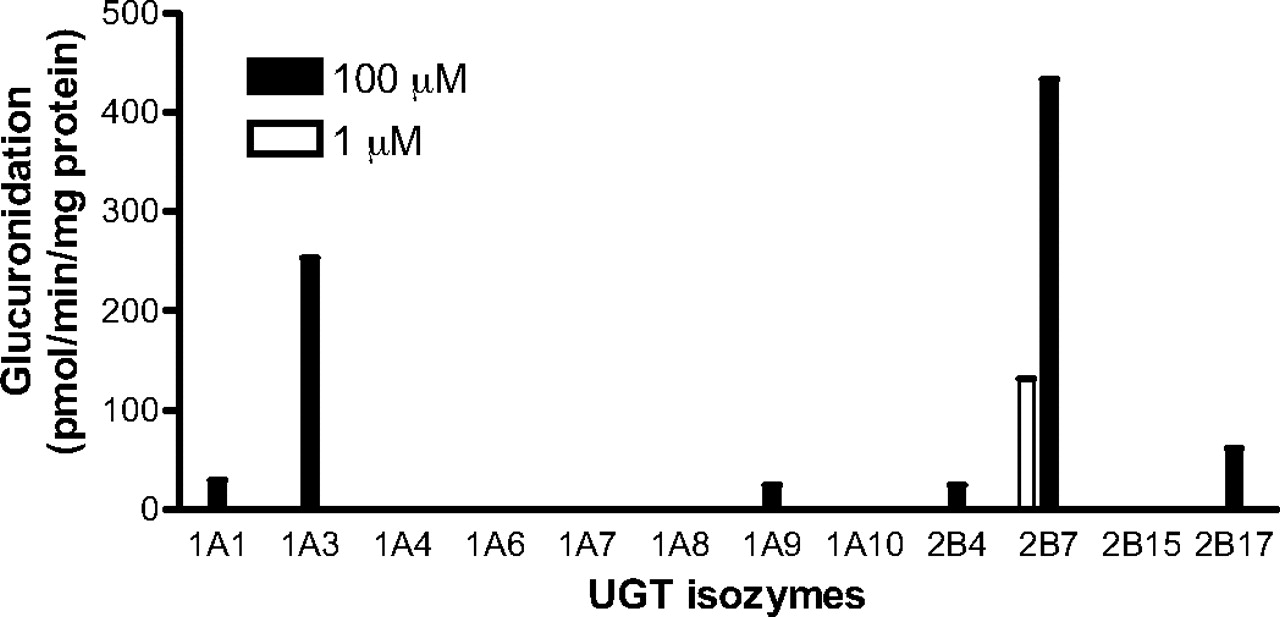
Gemfibrozil glucuronidation by recombinant human UGTs. Gemfibrozil was incubated with recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 for 20 min. Substrate concentrations of 1 and 100 μM and respective protein concentrations of 0.1 and 0.05 mg of protein/ml were used. The lower quantitation limit for the assay was 10 pmol/min/mg protein. Data represent the mean of duplicate determination.
Glucuronidation by Recombinant UGTs. At the therapeutic plasma concentration range of unbound gemfibrozil (1 μM), only UGT2B7 showed gemfibrozil glucuronidation activity. At 100 μM, UGT1A1, UGT1A3, UGT1A9, UGT2B4, and UGT2B17, as well as UGT2B7, possessed glucuronidation activity (Fig. 3). Glucuronidation velocities at 100 μM gemfibrozil for UGT1A1, UGT1A3, UGT1A9, UGT2B4, UGT2B7, and UGT2B17 were 29, 254, 25, 24, 433, and 62 pmol/min/mg protein, respectively, whereas the other recombinant UGT isozymes showed a velocity of less than 10 pmol/min/mg protein.
Further experiments to characterize the kinetics for gemfibrozil glucuronidation revealed Michaelis-Menten kinetics for recombinant UGT2B7 and UGT1A3. The Km values for UGT2B7 and UGT1A3 were 2.2 ± 0.3 and 696 ± 98 μM, respectively. The respective Vmax values were 353 ± 8.1 and 2829 ± 209 pmol/min/mg protein (Fig. 4, mean ± computer-calculated S.E.).
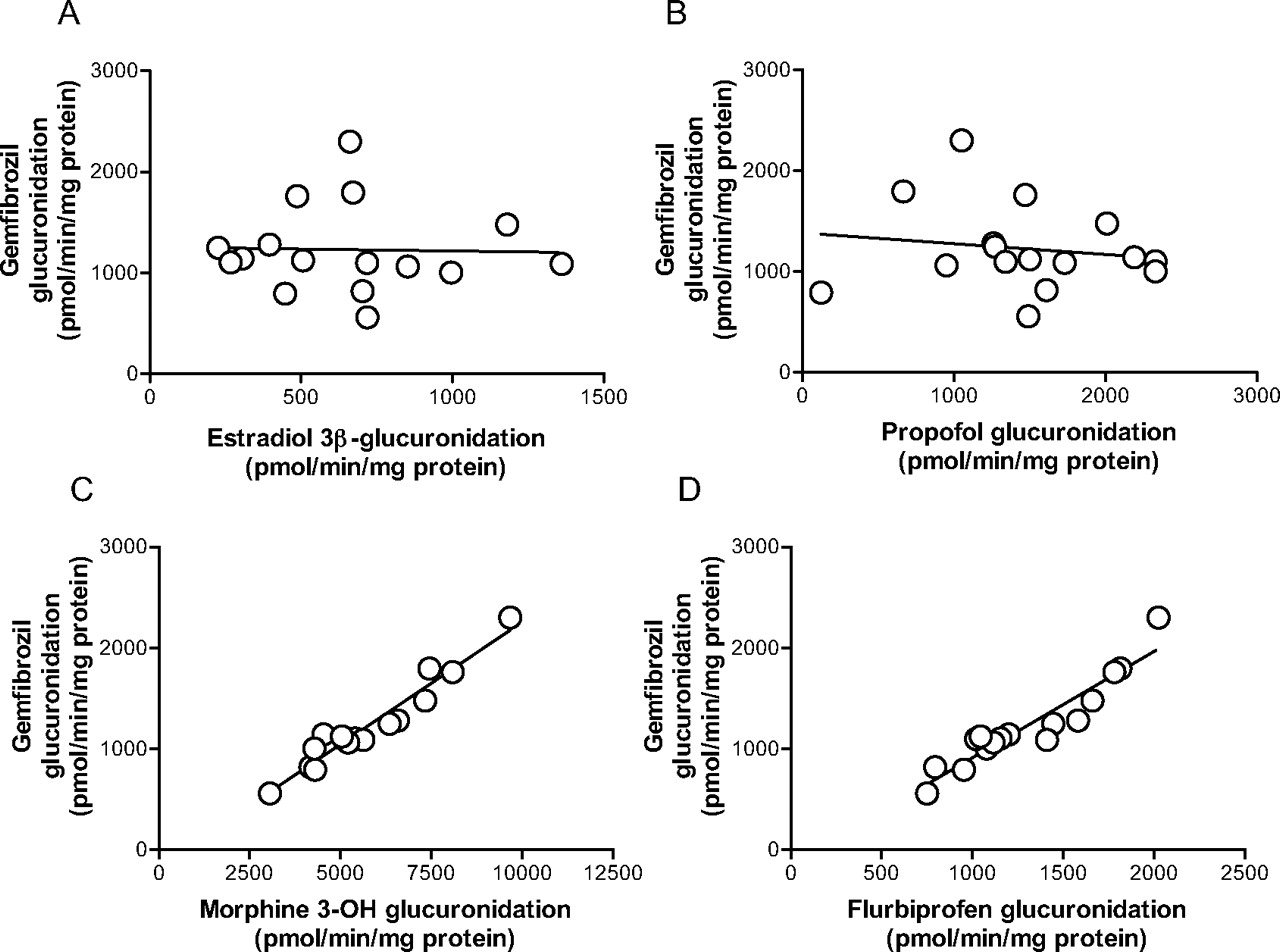
Correlation Study in HLMs. The glucuronidation velocity of gemfibrozil in microsomes from 16 human livers ranged from 559 to 2301 pmol/min/mg protein. As shown in Fig. 5, C and D, a significant correlation was found between gemfibrozil glucuronidation and the glucuronidation of both morphine at the 3-OH position (r = 0.966, p < 0.0001) and flurbiprofen (r = 0.937, p < 0.0001), two typical reactions for UGT2B7. In contrast, no significant correlation was found between gemfibrozil glucuronidation and estradiol 3β-glucuronidation (Fig. 5A) or propofol glucuronidation (Fig. 5B), two probe reactions for UGT1A1 and UGT1A9, respectively.
Chemical Inhibition in HLMs. The inhibitory effects of mefenamic acid and flurbiprofen on gemfibrozil glucuronidation in pooled HLMs were evaluated. As shown in Fig. 6, gemfibrozil glucuronidation was inhibited by mefenamic acid and flurbiprofen in a concentration-dependent manner. The IC50 values for mefenamic acid and flurbiprofen were calculated to be 1.0 ± 0.1 and 23 ± 3.4 μM, respectively (mean ± computer-calculated S.E.).
Discussion
We identified the main UGT isozymes responsible for gemfibrozil glucuronidation in the human liver by first investigating this reaction in pooled HLMs. The Eadie-Hofstee plot shows that gemfibrozil glucuronidation follows typical Michaelis-Menten kinetics with high and low affinity components (Fig. 2). Intrinsic clearance of the high affinity component contributes to 91% of the intrinsic clearance value, suggesting that this component is the predominant contributor to gemfibrozil glucuronidation in HLMs. A previous study showed that the total plasma concentration of gemfibrozil ranged from 15 to 25 μg/ml (60-100 μM) after twice-daily administration of 600 mg of gemfibrozil (Todd and Ward, 1988). Because gemfibrozil is extensively (97%) bound to serum albumin in vitro (Todd and Ward, 1988), plasma concentration of unbound gemfibrozil was calculated to be from 1.8 to 3 μM. This suggests that the high affinity component of the UGT isozyme plays a major role at the therapeutic plasma concentration of unbound gemfibrozil.
Gemfibrozil glucuronidation by recombinant human UGT isozymes showed that only UGT2B7 has detectable glucuronidation activity at 1 μM gemfibrozil (Fig. 3), whereas UGT1A1, UGT1A3, UGT1A9, UGT2B4, and UGT2B17 show glucuronidation activity at a higher concentration (100 μM) (Fig. 3). This finding is partly consistent with a previous report in which UGT1A1, UGT1A3, UGT1A9, UGT2B7, and UGT2B15 showed glucuronidation activity at a concentration of 250 μM (Prueksaritanont et al., 2002a). The discrepancy in results between the two studies may be attributable to differences in incubation conditions.
Because UGT1A3 and UGT2B7 showed relatively higher gemfibrozil glucuronidation activity (Fig. 3), we conducted a kinetic study for glucuronidation in recombinant UGT1A3 and UGT2B7. The kinetic profiles of recombinant UGT1A3 and UGT2B7 followed Michaelis-Menten kinetics (Fig. 4). The Km value for UGT2B7 was 2.2 μM, which is similar to the high affinity component in HLMs (2.5 μM), whereas the Km value for UGT1A3 was much higher (696 μM). To assess the contribution of UGT2B7 to gemfibrozil glucuronidation, a correlation analysis was carried out between gemfibrozil glucuronidation and both morphine 3-OH glucuronidation (Stone et al., 2003; Miners et al., 2006; Mano et al., 2007a) and flurbiprofen glucuronidation (Mano et al., 2007b), two reactions mainly catalyzed by UGT2B7, using microsomes prepared from 16 individual human livers. A significant correlation with minimal intercept in the correlation lines was observed between gemfibrozil glucuronidation and both morphine 3-OH glucuronidation and flurbiprofen glucuronidation (Fig. 5, C and D). However, no significant correlation was observed when comparing gemfibrozil glucuronidation with estradiol 3β-glucuronidation or propofol glucuronidation (Fig. 5, A and B), two typical reactions for UGT1A1 and UGT1A9, respectively. Although the relative abundance of UGT2B7 in the liver relative to other UGTs remains to be investigated, these findings suggest that the contribution of UGT isozymes other than UGT2B7 is small, and that UGT2B7 is the main UGT isozyme responsible for gemfibrozil glucuronidation in the human liver. A correlation study in HLMs with a probe substrate for UGT1A3 is useful for eliminating this isozyme as an important contributor to gemfibrozil glucuronidation. However, the probe substrate for UGT1A3 is not fully understood, and only F6-1α, 23S,25-trihydroxyvitamin D3 has been reported as a possible probe substrate for this isozyme (Kasai et al., 2005), which is not a commercially available substrate. For this reason, a correlation study using a probe reaction for UGT1A3 could not be conducted. Because it has been reported that the mRNA level of UGT1A3 was approximately 20-fold less than that of UGT1A1 (Mojarrabi et al., 1996), it is unlikely that UGT1A3 would play a significant role in gemfibrozil glucuronidation.
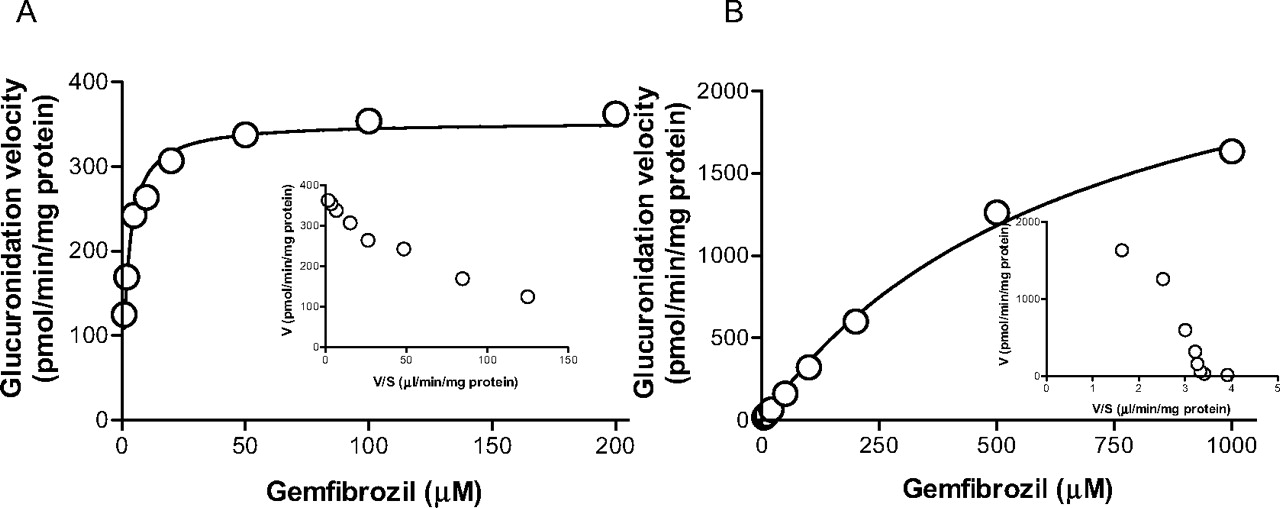
Kinetics of gemfibrozil glucuronidation by recombinant UGT1A3 and UGT2B7. A, gemfibrozil (1-200 μM) was incubated with UGT2B7 (0.05 mg of protein/ml) for 15 min. B, gemfibrozil (5-1000 μM) was incubated with UGT1A3 (0.1 mg of protein/ml) for 15 min. The Eadie-Hofstee plot is shown as an inset. Data represent the mean of duplicate determination.

Correlation between gemfibrozil glucuronidation and other glucuronosyltransferase activities in microsomes from 16 human livers. Gemfibrozil (10 μM) was incubated with individual HLMs (0.1 mg of protein/ml) for 10 min. Estradiol 3β-glucuronidation (A), propofol glucuronidation (B), morphine 3-OH glucuronidation (C), all typical reactions for UGT1A1, UGT1A9, and UGT2B7, respectively, were provided by the manufacturer. Flurbiprofen glucuronidation (D), which is predominantly catalyzed by UGT2B7, is based on a previous report (Mano et al., 2007). Data represent the mean of duplicate determination.
It is valuable to determine the inhibitory potentials of gemfibrozil against UGTs to evaluate possible drug-drug interactions via glucuronidation. The main purpose of this study is to determine the UGT isozymes responsible for gemfibrozil glucuronidation. For that reason, gemfibrozil's effects on UGTs need further investigation. However, some reports about gemfibrozil's effects on UGTs are available. Goosen et al. (2007) have recently reported that gemfibrozil inhibited atorvastatin lactonization and atorvastatin lactone glucuronidation with IC50 values of 346 and >1000 μM, respectively. The highest rates of lactonization or atorvastatin lactone glucuronidation were mediated by UGT1A1 and UGT1A3, or UGT1A3 and UGT1A4, respectively (Goosen et al., 2007). The study by Prueksaritanont et al. (2002b) showed that gemfibrozil inhibited simvastatin glucuronidation (IC50 = 354 μM); simvastatin is mainly glucuronidated by UGT1A1 and UGT1A3. These findings suggest that the inhibitory potential of gemfibrozil for UGT1A3 is low.

Inhibitory effects of mefenamic acid and flurbiprofen on gemfibrozil glucuronidation in HLMs. Gemfibrozil (1 μM) was incubated with pooled HLMs (0.1 mg of protein/ml) for 10 min in the absence and presence of mefenamic acid (0.1-10 μM) or racemic flurbiprofen (1-100 μM). Data represent the mean of duplicate determination.
A separate inhibition study showed similar IC50 values of mefenamic acid for gemfibrozil glucuronidation in HLMs (1.0 μM) and UGT2B7-catalyzed flurbiprofen glucuronidation in recombinant UGT2B7 (0.73 or 1.7 μM) (Mano et al., 2007b). Flurbiprofen also showed potent inhibition of gemfibrozil glucuronidation in HLMs (IC50,23 μM) (Fig. 6). The IC50 value of flurbiprofen in HLMs is also similar to the value reported for (S)-flurbiprofen in UGT2B7-catalyzed gemcabene glucuronidation (27.4 μM) (Bauman et al., 2005). Because the IC50 values of (S)-flurbiprofen in UGT1A3- and UGT2B17-catalyzed gemcabene glucuronidation are >1000 and 653 μM, respectively (Bauman et al., 2005), the contribution of these two UGT isozymes to gemfibrozil glucuronidation is not considered significant. These findings also show that the UGT2B7 isozyme plays a predominant role in the glucuronidation of gemfibrozil.
UGT2B7 is capable of conjugating a wide variety of xenobiotics and endogenous steroids. Substrates include androsterone, morphine, codeine, lorazepam, propranolol, and nonsteroidal anti-inflammatory drugs (de Wildt et al., 1999). UGT2B7 is an inducible enzyme (Munzel et al., 1999) and has more than 10 single nucleotide polymorphisms (Holthe et al., 2003). Identification of the UGT2B7 isozyme responsible for gemfibrozil glucuronidation will prove to be an invaluable tool in identifying the impact of UGT2B7 polymorphisms on the variability associated with gemfibrozil pharmacokinetics and magnitude of drug-drug interactions.
Footnotes
-
doi:10.1124/dmd.107.017269.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; HLM, human liver microsome.
- Received June 14, 2007.
- Accepted July 31, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}