


Abstract
Mycophenolic acid (MPA) is an immunosuppressive agent commonly used after organ transplantation. Altered concentrations of MPA metabolites have been reported in diabetic kidney transplant recipients, although the reason for this difference is unknown. We aimed to compare MPA biotransformation and UDP-glucuronosyltransferase (UGT) expression and activity between liver (n = 16) and kidney (n = 8) from diabetic and nondiabetic donors. Glucuronidation of MPA, as well as the expression and probe substrate activity of UGTs primarily responsible for MPA phenol glucuronide (MPAG) formation (UGT1A1 and UGT1A9), and MPA acyl glucuronide (AcMPAG) formation (UGT2B7), was characterized. We have found that both diabetic and nondiabetic human liver microsomes and kidney microsomes formed MPAG with similar efficiency; however, AcMPAG formation was significantly lower in diabetic samples. This finding is supported by markedly lower glucuronidation of the UGT2B7 probe zidovudine, UGT2B7 protein, and UGT2B7 mRNA in diabetic tissues. UGT genetic polymorphism did not explain this difference because UGT2B7*2 or *1c genotype were not associated with altered microsomal UGT2B7 protein levels or AcMPAG formation. Furthermore, mRNA expression and probe activities for UGT1A1 or UGT1A9, both forming MPAG but not AcMPAG, were comparable between diabetic and nondiabetic tissues, suggesting the effect may be specific to UGT2B7-mediated AcMPAG formation. These findings suggest that diabetes mellitus is associated with significantly reduced UGT2B7 mRNA expression, protein level, and enzymatic activity of human liver and kidney, explaining in part the relatively low circulating concentrations of AcMPAG in diabetic patients.
Introduction
Mycophenolic acid (MPA) is an immunosuppressive agent widely used to prevent rejection following organ transplantation. Most of the administered dose (87–94%) ultimately appears in the urine as the pharmacologically inactive phenolic 7-O-glucuronide of MPA (MPAG) with small percentages reported to be biotransformed to either pharmacologically active acyl glucuronide (AcMPAG) or inactive glucoside conjugates (Shipkova et al., 1999). It has been suggested that AcMPAG may be the culprit for some of the adverse side effects of MPA, including gastrointestinal (GI) toxicity (Wieland et al., 2000). MPA exhibits prominent pharmacokinetic features consisting of a secondary peak observed in the MPA concentration-time profile. The latter is considered to result from hepatic MPA glucuronidation, followed by biliary excretion, hydrolysis in the intestines to MPA, and subsequent reabsorption of parent MPA. This drug and its metabolites are also transported by organic anion transporters, organic anion-transporting polypeptide, and by multidrug resistance-associated protein 2 (Barraclough et al., 2010).
The uridine-5′-diphosphate-glucuronosyltransferases (UGTs) are a superfamily of membrane-bound enzymes that catalyze glucuronidation at nucleophilic functional groups in xenobiotics and endogenous compounds, leading to the formation of more hydrophilic derivatives for excretion in bile and/or urine. UGTs are classified based on the similarity in gene sequence. So far, all UGTs involved in the metabolism of marketed drugs are originated from the UGT 1A, 2A, and 2B subfamilies and include 19 distinct catalytically active UGTs in humans (Mackenzie et al., 2005). Various studies have used recombinantly expressed enzymes to identify the specific UGT enzymes involved in the glucuronidation of MPA, with some disagreement among reports. Mackenzie (2000) initially reported that UGTs 1A8, 1A9, and 1A10 were capable of forming MPAG using enzymes transiently expressed in COS-7 cells. However, the authors also reported a lack of detectable MPA glucuronidation activity for UGTs 1A1, 1A3, 1A6, 2B4, and 2B7. Shipkova et al. (2001) then reported that recombinant UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A10, 2B4, 2B7, and 2B15 expressed in insect cells were all capable of forming MPAG. However, Basu et al. (2004) using COS-1 expressed UGTs and Bernard and Guillemette (2004) using HEK293 expressed UGTs suggested that UGT1A7, 1A8, 1A9, and 1A10 were the main UGTs that could produce MPAG. Finally, a study by Picard et al. (2005) suggested that UGT1A9 and 1A10 are the major contributors to MPAG production. These discrepancies in data could result from different experimental conditions and cell systems used for UGT expression. However, taking into consideration all studies published so far, it appears that UGT1A9, which is mainly expressed in liver and kidney, and UGT1A10, which is mainly expressed in intestines, may be the main enzymes responsible for phenolic glucuronidation of MPA. Although less studied, UGT2B7, which is mainly expressed in liver, kidney, and intestines, appears to be the most important UGT involved in the production of AcMPAG (Bernard et al., 2006).
The pharmacokinetics of MPA and its metabolites show high variability in various transplant subpopulations (Ensom et al., 2002). In previous clinical pharmacokinetics studies in kidney transplant recipients, we have described significantly higher MPAG/AcMPAG plasma concentration ratios in diabetic versus nondiabetic patients (Akhlaghi et al., 2006; Patel et al., 2007). This observation suggests that diabetes may influence UGT enzymes responsible for the formation of MPAG and/or AcMPAG or may alter other mechanisms governing the circulating concentration of these metabolites. However, the underlying molecular mechanisms are not yet known.
The purpose of the current study was to determine whether diabetes mellitus is associated with altered rates of microsomal MPAG and/or AcMPAG formation ex vivo using liver and kidney microsomes obtained from both diabetic donors and nondiabetic controls. We also evaluated whether the identified differences were correlated with differences in the activity and expression of selected UGTs involved in the biotransformation of MPA to MPAG (UGT1A9) or to AcMPAG (UGT2B7).
Materials and Methods
Chemicals.
AcMPAG, MPA, and MPAG were donated by Roche Pharmaceuticals (Palo Alto, CA). 3-Acetamidophenol, estradiol, acetophenetidin (phenacetin), phenolphthalein glucuronic acid, propofol, and thymol were purchased from Sigma-Aldrich (St. Louis, MO). Zidovudine (AZT), AZT-glucuronide, estradiol-3-β-glucuronide, estradiol-17-β-glucuronide, and propofol-glucuronide were purchased from TRC (Toronto, ON, Canada), whereas high-performance liquid chromatography (HPLC)-grade acetonitrile and methanol were obtained from Pharmco Products, Inc. (Brookefield, CT). All other reagents and solvents were obtained from general commercial suppliers. All chemicals were used without further purification.
In Vitro Study.
Tissues and microsomal preparation.
Diabetic and nondiabetic human liver and kidney samples were obtained from XenoTech, LLC (Lenexa, KS). The tissues were selected to match, as best as possible, according to demographic characteristics between diabetic and nondiabetic donors (Table 1). Microsomal fractions were prepared as described previously (Guengerich and Bartleson, 2001) and were stored at −80°C until use. Protein concentrations were estimated using a bicinchoninic acid method (Pierce-Fisher, Rockford, IL). To measure the extent of oxidative stress, malondialdehyde, a product of lipid peroxidation, was measured in tissues using thiobarbituric acid assay (Ernster and Nordenbrand, 1967). The fraction of drug unbound in the microsomal fraction was determined by equilibrium dialysis against 0.1 M phosphate buffer (pH 7.4) using Slide-A-Lyzer Dialysis Cassettes, 2000 MWCO, 0.5 ml, according to the manufacturer's manual (Pierce-Fisher).
Demographic characteristics of human liver and kidney samples from diabetic and nondiabetic donors
UGT Western blot analyses.
Amounts of UGT2B7 in human liver microsomes (HLMs) and human kidney microsomes (HKMs) were determined by quantitative Western immunoblotting by modifying a method used for cytochrome P450 as described previously (Guengerich et al., 1982). Rabbit anti-human UGT2B7 antisera with recombinant UGT2B7 standard were obtained from BD Biosciences (San Jose, CA). Protein levels were measured in duplicate, and mean values were used for quantification. Expressed UGT2B7 (BD Biosciences) was used as a reference standard. Known amounts of expressed UGT2B7 characterized by 7-hydroxy-4-trifluoromethylcoumarin [pmol/(min × mg protein)] were used on each gel and the results were quantified using densitometry. Each sample was quantified separately, based on the concentration curve, and used for statistical analysis.
UGT activity assays.
Incubations (100 μl total volume) were performed in 50 mM phosphate buffer (pH 7.4) at 37°C, 4.0 mM MgCl2, 4.0 mM uridine diphosphoglucuronic acid, and alamethicin (50 μg/mg protein). Microsomal fractions were preincubated on ice for 15 min with alamethicin, which was found to maximally activate microsomal UGT activity. Probes and MPA were added to the reaction in 5% (v/v) final methanolic concentration. No change in the UGT2B7 activity was observed after the addition of 5% (v/v) methanol. After equilibrating incubation tubes at 37°C for 5 min, reactions were initiated by the addition of MgCl2 and uridine diphosphoglucuronic acid and were allowed to proceed at 37°C for 90 min in a shaking water bath. Reactions were stopped by the addition of 100 μl of ice-cold acetonitrile with internal standard. Tubes were placed on ice for 10 min and then centrifuged at 10,000g for 10 min at 4°C to remove the precipitate. The supernatant was transferred to HPLC tubes and partially dried down in a centrifugal evaporator at room temperature before analysis by HPLC. Acidification of acyl-glucuronides is very important for postreaction handling (Shipkova et al., 2000). However, no differences in rate of change were found when microsomal incubations with and without acidification from diabetic and nondiabetic subjects were compared.
HPLC LC-MS/MS quantitation.
MPA and metabolites were quantified by HPLC-UV as described previously (Patel and Akhlaghi, 2006). HPLC assays used for quantification of estradiol (UGT1A1), propofol (UGT1A9), and their glucuronide metabolites have been described in detail previously (Court, 2005). The chromatographic separation was performed on an HPLC Hitachi D-7000 series instrument (Hitachi, San Jose, CA). Data from the detector were collected and analyzed with HPLC system manager for Hitachi D-7000 software. An assay for quantification of AZT (UGT2B7) and its glucuronide metabolite was described previously (Engtrakul et al., 2005). Chromatography was performed on an liquid chromatography-tandem mass spectrometry that included a binary pump and autosampler (Shimadzu, Kyoto, Japan) coupled to an AB Sciex triple quadrupole mass spectrometric detector API 3200 (AB Sciex, Toronto, ON, Canada), equipped with Turbo V source electrospray ionization probe. The column was heated to 50°C using Flatron Systems TC-50 temperature controller and CH-30 column heater (ASTEC, Whippany, NJ). The chromatographic data were collected and analyzed using Analyst package (version 1.4.1.; AB Sciex). Calculated rates of formation of MPAG, AcMPAG, AZT-glucuronide, 3β- and 17β-estradiol glucuronides, and propofol glucuronide were normalized to incubation time and total protein content.
Quantitative reverse transcription-polymerase chain reaction.
Total RNA was isolated with RNA-Bee system (Tel-Test Inc., Friendswood, TX), according to the manufacturer's manual and as described previously (Yang et al., 2007). Levels of UGT1A1, UGT1A9, and UGT2B7 mRNAs were determined by real-time polymerase chain reaction (PCR) (model 7300; Applied Biosystems, Foster City, CA) with SYBR Green detection as previously described with minor modifications (Manevski et al., 2010). Quantitative PCRs (25 μl) included SYBR Green 2× Master Mix (Applied Biosystems), 10 μl of diluted cDNA, and 100 nM of each primer (200 nM for 18S RNA). Primer pair sequences were as follows: CCC CTC GAT GCT CTT AGC TGA GTG T (18S-rRNA-forward), CGC CGG TCC AAG AAT TTC ACC TCT (18S-rRNA-reverse), GCT TTT GTC TGG CTG TTC CCA CT (UGT1A1-forward), TCG AAG GTC ATG TGA TCT GAA TGA GA (UGT1A1-reverse), GGA GCC ACT GGT TCA CCA TGA G (UGT1A9-forward), AGA TCC TCC AGG GTA TAT GAA GTT GAA (UGT1A9-reverse), and TTT CAC AAG TAC AGG AAA TCA TGT CAA T (UGT2B7-forward), CAG CAG CTC ACT ACA GGG AAA AAT (UGT2B7-reverse).
UGT2B7 genotyping.
Liver samples were genotyped for single nucleotide polymorphisms in the UGT2B7 exon 2 region by direct sequencing of genomic DNA as described previously (Court et al., 2003). Single nucleotide polymorphisms included rs7439366 (c.802C>T; H268Y; UGT2B7*2) and rs28365062 (c.735A>G; UGT2B7*1c) that have been previously associated with altered AZT glucuronidation in vitro and in vivo (Kwara et al., 2009).
Enzyme kinetic data analysis.
Data were fitted to appropriate models by nonlinear least-squares regression using GraphPad Prism (version 5.00 for Windows; GraphPad Software Inc., San Diego, CA). For AcMPAG and MPAG formation from MPA or AZT-glucuronide formation from AZT, a model incorporating substrate inhibition equation [Y = Vmax · S/(Km+S · (1+S/Ksi))] was developed, with S representing the concentration of substrate. The estimated parameters were maximum rate of formation (Vmax) and Michaelis-Menten constant (Km). Uncompetitive substrate inhibition constant (Ksi) was fixed to a nominal value for each substrate, and intrinsic clearance was calculated as Vmax/Km.
Statistical analysis.
All statistical analyses were performed with SPSS (version 16; SPSS Inc., Chicago, IL). The Kolmogorov-Smirnov test was used to test for normal distribution of data. The differences between diabetic and nondiabetic groups were determined using the Student's t test, and P < 0.05 was considered to be statistically significant. All data are expressed as mean ± S.E.
Results
Demographic data for the liver (n = 16) and kidney (n = 8) donors are presented in Table 1. For livers, there were no differences between the two groups with respect to age, gender, ethnicity, and liver fat content (Table 1). Kidney samples were slightly dissimilar with respect to donor gender and ethnicity (Table 1).
Increased oxidative stress is a widely accepted contributor in the development and progression of diabetes mellitus. Diabetes is usually accompanied by an increased production of free radicals, thus increasing oxidative stress in the body. The rate of formation of thiobarbituric acid reactive substances (TBARSs) was measured as a marker of diabetes-associated oxidative stress. Formation of TBARSs was significantly higher in HLMs (P < 0.001) but not in HKMs (P = 0.053) from diabetic donors (Table 1).
MPA Glucuronidation by Diabetic and Nondiabetic Liver and Kidney.
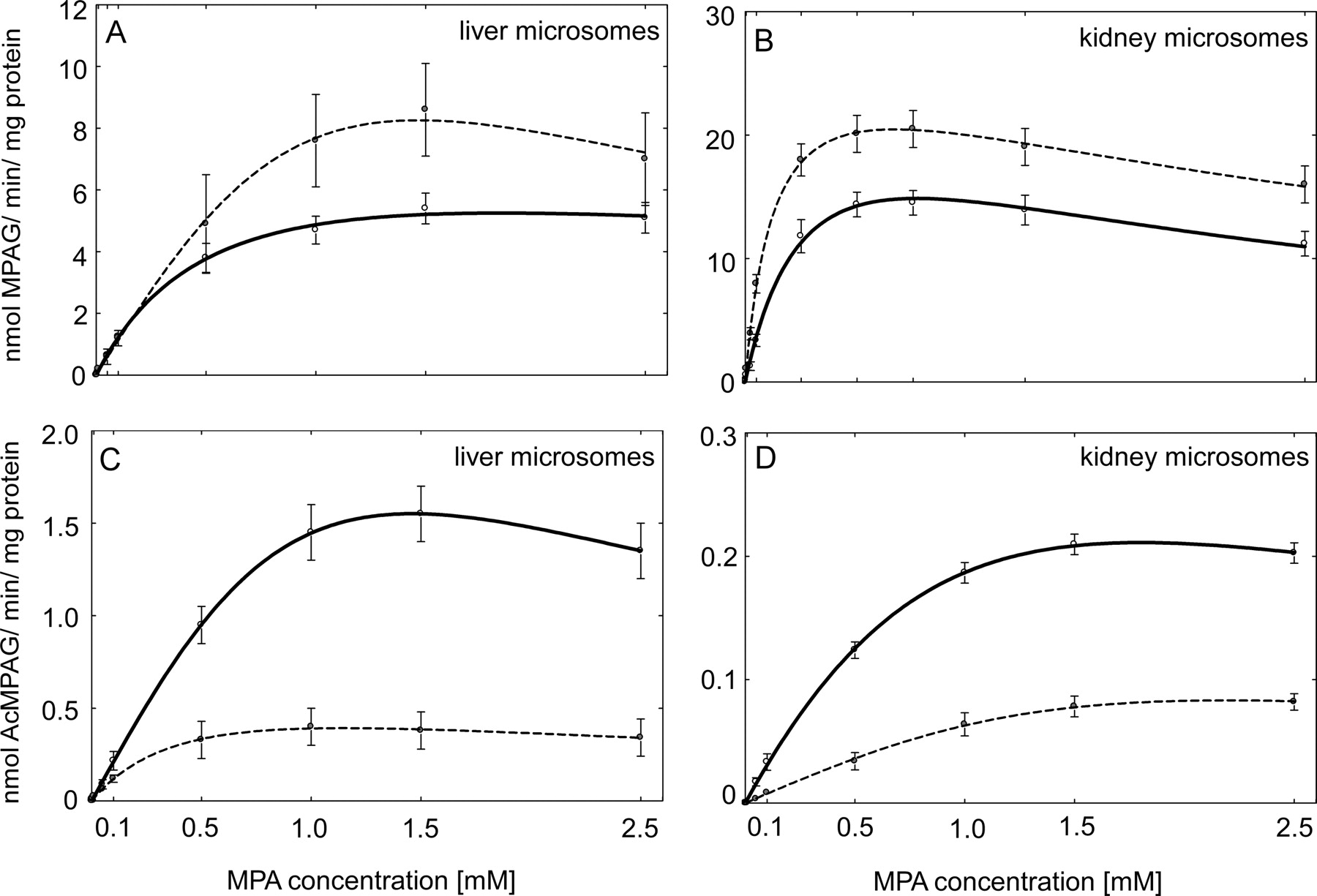
To characterize MPA-glucuronidation activities of HLMs and HKMs, individual liver and kidney samples were assayed for MPA UGT activity. MPA shows negligible binding to HLMs, because the ratios of MPA concentration in the buffer and microsomal compartments of dialysis cassette were 1.02:1, 0.96:1, and 0.97:1 for diabetic and 1.04:1, 0.99:1, and 0.97:1 for nondiabetic HLM samples for added 10.0 μM, 100 μM, and 1 mM MPA, indicating an absence of nonspecific binding. All microsomal preparations from liver and kidney samples, either from diabetic or nondiabetic donors, were capable of forming both MPAG and AcMPAG. MPAG and AcMPAG formation followed substrate inhibition kinetics in both tissues (Fig. 1). The kinetic constants for the two metabolites are summarized in Table 2. It is interesting to note that the Vmax for MPAG formation was higher with nondiabetic HKMs than with HLMs from nondiabetic donors. However, the Km values were similar for these microsomal preparations. Extrapolated whole organ Vmax/Km values were estimated at 31.3 ± 9.9 for liver (40 mg · g−1, organ weight is 1500 g) and 38.8 ± 2.6 for kidney (6 mg · g−1, organ weight is 300 g). Thus, the formation of MPAG occurs predominantly in the liver tissue. Furthermore, kidney microsomes were more capable than liver microsomes to form AcMPAG because the Km value was significantly higher for nondiabetic HKMs than with nondiabetic HLMs (P < 0.001).

Formation rate of MPAG and AcMPAG from MPA in diabetic and nondiabetic HLM (eight in each group) and HKM (four in each group) samples. The rate of formation of liver MPAG (A) and AcMPAG (C) and kidney MPAG (B) and AcMPAG (D) when HLM or HKM samples from nondiabetic (solid line) and diabetic (dashed line) donors incubated with MPA. All data are expressed as mean ± S.E.
Enzyme kinetic parameters of the formation of MPAG and AcMPAG from MPA
Apparent Km, Vmax, and Ksi values for the formation of metabolites when MPA was incubated with diabetic and nondiabetic HLMs and HKMs, respectively.
UGT2B7, UGT1A1, and UGT1A9 Probe Activities in Diabetic and Nondiabetic Liver and Kidney.
To assess the effect of diabetes on liver and kidney UGT2B7 activity, we examined the rate of AZT-glucuronidation. Rates of AZT glucuronidation were significantly lower (P < 0.001) in both HLMs (Table 3; Fig. 2D) and HKMs (Table 3; Fig. 3D) from diabetic donors than those from nondiabetic donors. Moreover, the correlation coefficient between AZT and AcMPAG Vmax values was 0.95, indicating a good agreement in UGT2B7 activity assessed using two markers. AZT shows negligible binding to HLM, free fraction for AZT was fu = 0.98, 0.96, and 0.99 for nondiabetic, and fu = 0.99, 0.94, and 0.96 for diabetic samples for added 1, 10, and 100 μM AZT, indicating an absence of nonspecific binding. To investigate the effect of diabetes on liver and kidney UGT1A1 and UGT1A9 activity, we examined the rate of 3β- and 17β-estradiol and propofol glucuronidation, respectively. Both estradiol (Fig. 5, C and D) and propofol (Fig. 6, C and D) glucuronidation rates were not significantly different in HLMs and HKMs from diabetic versus nondiabetic donors.
Enzyme kinetic parameters of the formation of AZT glucuronide
Apparent Km and Vmax for the formation of metabolites when AZT was incubated with diabetic and nondiabetic HLMs and HKMs, respectively.

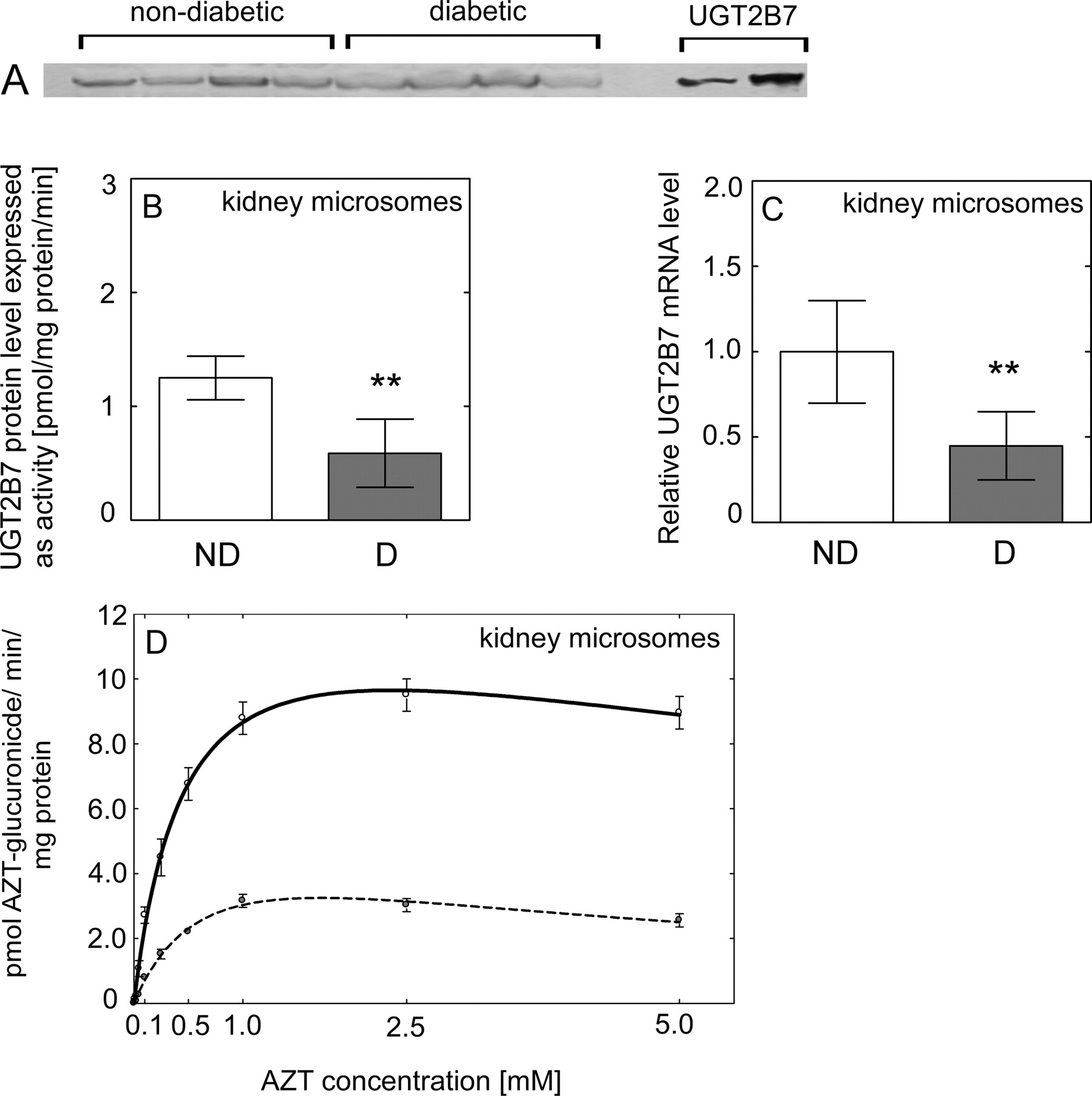
Effect of diabetes on UGT2B7 expression and activity in human liver. A, representative Western blot of UGT2B7 protein level in nondiabetic and diabetic liver samples. B, UGT2B7 protein level in nondiabetic (ND, n = 8) (□) and diabetic (D, n = 8) (■) HLMs quantified based on recombinant 7-hydroxy-4-trifluoromethylcoumarin glucuronidation activity (see Materials and Methods). C, the level of UGT2B7 mRNA expressed relative to that in nondiabetic (considered as 1). D, the rate of formation of AZT glucuronide when HLMs from nondiabetic (solid line) and diabetic (dashed line) donors incubated with AZT. All data are expressed as mean ± S.E and statistical significance related to a control group (***, P < 0.001; **, P < 0.01).

Effect of diabetes on UGT2B7 expression and activity in human kidney. A, representative Western blot of UGT2B7 protein level in nondiabetic and diabetic kidney samples. B, UGT2B7 protein level in nondiabetic (ND, n = 4) (□) and diabetic (D, n = 4) (■) HKMs quantified based on recombinant 7-hydroxy-4-trifluoromethylcoumarin glucuronidation activity (see Materials and Methods). C, the level of UGT2B7 mRNA expressed relatively to this in nondiabetic (considered as 1). D, the rate of formation of AZT glucuronide when HKMs from nondiabetic (solid line) and diabetic (dashed line) donors incubated with AZT. All data are expressed as mean ± S.E and statistical significance related to a control group (**, P < 0.01).
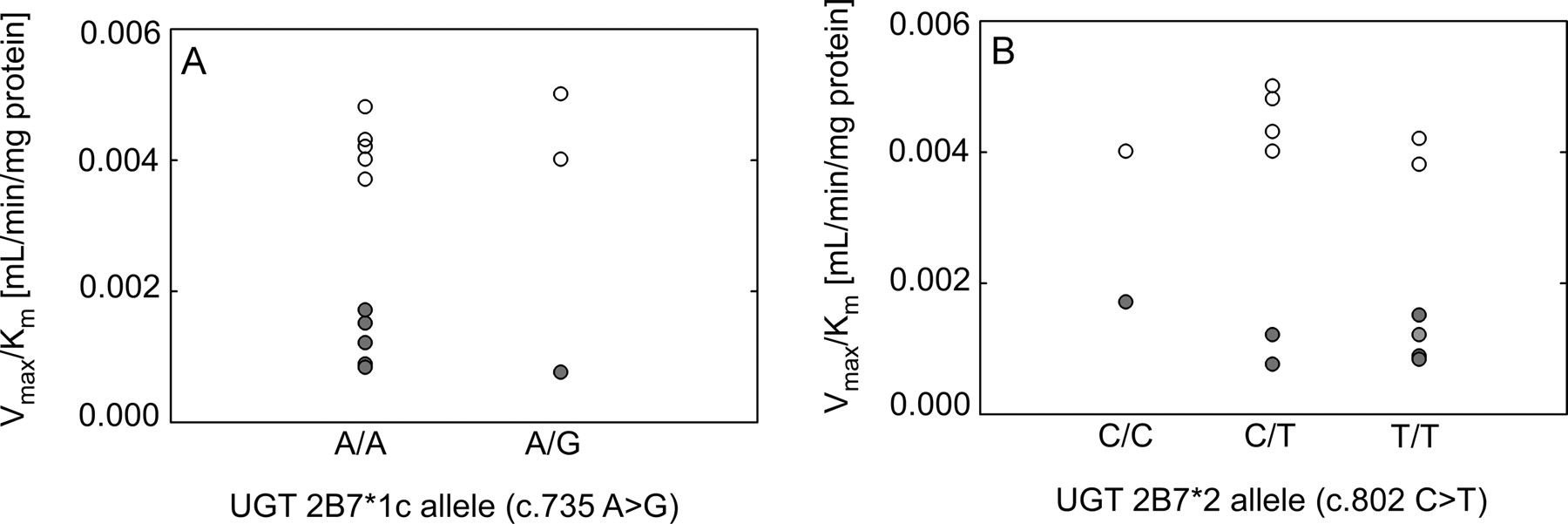
Effect of liver UGT2B7 genotype on AcMPAG formation. Effect of liver UGT2B7*1c allele (c.735 A>G) (A) and UGT2B7*2 allele (c.802 C>T) (B) on AcMPAG formation in HLMs from nondiabetic (○) and diabetic donors (●). Values of intrinsic clearance (Vmax/Km) for each liver are shown.

Effect of diabetes on liver and kidney UGT1A1 mRNA and activity. UGT1A1 mRNA level in liver (n = 8) (A) and kidney (n = 4) (B) samples from nondiabetic (ND, □) and diabetic (D, ■) donors expressed relative to that in nondiabetic (considered as 1). The rate of formation of estradiol 3-β-glucuronide (3-β-EDG) (mediated primarily by UGT1A1) and estradiol 17-β-glucuronide (17-β-EDG) (mediated in part by UGT2B7) in HLMs (C) and HKMs (D) from nondiabetic (ND, □) and diabetic (D, ■) donors incubated with estradiol. All data are expressed as mean ± S.E and statistical significance related to a control group (#, nonsignificant).

Effect of diabetes on liver and kidney UGT1A9 mRNA and activity. UGT1A9 mRNA level in liver (n = 8) (A) and kidney (n = 4) (B) samples from nondiabetic (□) and diabetic (■) donors expressed relative to that in nondiabetic (considered as 1). The rate of formation of propofol glucuronide (probe for UGT1A9) when HLMs (C) and HKMs (D) from nondiabetic (□) and diabetic (■) donors were incubated with propofol. All data are expressed as mean ± S.E.
UGT2B7 Protein Content of Diabetic and Nondiabetic Liver and Kidney.
Immunoquantitation of UGT2B7 protein content in microsomes revealed significantly lower UGT2B7 levels in diabetic HLMs (P < 0.001) (Fig. 2B) and HKMs (P = 0.009) (Fig. 3B) than those from nondiabetic donors.
UGT2B7, UGT1A1, and UGT1A9 mRNA Levels in Diabetic and Nondiabetic Liver and Kidney.
The level of UGT2B7 mRNA was significantly lower in diabetic liver (P = 0.001) (Fig. 2C) and kidney (P = 0.034) (Fig. 3C). The mRNA level of both UGT1A1 (Fig. 5, A and B) and UGT1A9 (Fig. 6, A and B) was not significantly different in diabetic liver or kidney compared with those from nondiabetic tissues, with large interindividual variability in both groups.
Influence of Liver UGT2B7 Genotype on AcMPAG Formation.
DNA extracted from liver tissue was genotyped for common UGT2B7 polymorphisms previously associated with altered UGT2B7 expression and activity. Genotyping for the UGT2B7*1c allele (c.735 A>G) identified 11 A/A homozygous individuals, 4 A/G heterozygous individuals, and 0 G/G homozygous individuals. Likewise, assay of the UGT2B7*2 allele (c.802 C>T) identified two C/C homozygous individuals, seven C/T heterozygous individuals, and six T/T homozygous individuals. Genotypes could not be determined in one liver sample. There were no differences in AcMPAG intrinsic clearance with respect to UGT2B7*1c (Fig. 4A) or UGT2B7*2 (Fig. 4B) genotypes.
Discussion
The liver is quantitatively the most important site of glucuronidation for most compounds (McGurk et al., 1998). The overall contribution of extrahepatic tissues to glucuronidation is generally lower than that of the liver (McGurk et al., 1998). However, several studies highlighted the fact that the kidney may significantly contribute to MPA biotransformation, based on comparison of urinary clearance of MPAG after oral or intravenous administration of mycophenolate mofetil and on the impact of hepatic impairment on MPAG clearance (Bullingham et al., 1996; Parker et al., 1996). Zucker et al. (1999) provided additional confirmation and reported that purified kidney extracts contained higher amounts of UGT enzymes involved in MPAG formation than those from liver samples. In our experiments, the HKMs were shown to have the highest intrinsic clearance for glucuronidation of MPA to its principal metabolite, MPAG. In contrast, the highest AcMPAG formation rate was found in HLMs. However, when these results extrapolated to the whole organ, observations from our ex vivo experiment suggest that liver is the organ primarily responsible for the systemic clearance of MPA, with some contribution from the kidney. Therefore, our data on MPA glucuronidation are similar to those published previously (Bowalgaha and Miners, 2001; Shipkova et al., 2001).
Previous clinical pharmacokinetic studies conducted by our group (Akhlaghi et al., 2006; Patel et al., 2007) reported a higher MPAG/AcMPAG concentration ratio (or lower AcMPAG/MPAG concentration ratio) in stable kidney transplant recipients with diabetes. In parallel to our clinical observations, MPAG concentration was not significantly different when MPA incubated with both HLM and HKM samples from diabetic donors. In our experiment, data from immunoblotting using a nonspecific antibody showed higher protein levels of UGT1A in diabetic HLMs than in nondiabetic HLMs (M. Dostalek, M. H. Court, and F. Akhlaghi, unpublished data). However, no differences were found in the activity of liver and kidney UGT1A9 using propofol as a specific probe. Moreover, MPAG is also formed by other UGTs present in liver tissue such as UGT1A1, 1A3, and 1A6 (Shipkova et al., 2001), and UGT1A1 was identified as a principal UGT underlying MPAG formation in rat liver (Miles et al., 2005). In our experiment, no change in UGT1A1 activity was observed in diabetic samples using estradiol as a specific probe.
The formation of AcMPAG was markedly lower in both HLM and HKM samples from diabetic donors. Using incubation with AZT, a specific substrate of UGT2B7 (Court et al., 2003), immunological blotting, and real-time PCR, we demonstrated that diabetes significantly decreases mRNA expression, protein level, and enzymatic activity of liver and kidney UGT2B7. However, the magnitude of decrease in ex vivo production of AcMPAG in diabetic tissue is much higher than the somewhat smaller changes in MPAG/AcMPAG concentration ratio observed in our clinical studies (Akhlaghi et al., 2006; Patel et al., 2007). At least two main explanations can be proposed to explain the discrepancy between in vitro data and data obtained from clinical studies. First, extrahepatic tissues might contain UGTs other than UGT2B7 capable of forming AcMPAG but are unaffected by diabetes. For example, UGT1A8, which is primarily expressed in extrahepatic tissue, is also capable of forming AcMPAG (Bernard et al., 2006). Second, the excretion of AcMPAG appears to depend on multidrug resistance-associated protein 2 (Westley et al., 2006), organic anion transporters, or other transporters that potentially can influence the pharmacokinetics of AcMPAG. For a better understanding of the underlying changes of AcMPAG in diabetic patients, elucidation of the expression of transporter proteins is warranted in diabetic tissues.
The impact of UGT2B7 polymorphism on the glucuronidation activity and on MPA exposure remains controversial (Kagaya et al., 2007). Previously published studies demonstrated that UGT2B7*2 polymorphism may affect UGT2B7 protein level or enzymatic activity (Barbier et al., 2000). However, various other studies have failed to show an effect of UGT2B7*2 on UGT2B7 expression or activity (Bhasker et al., 2000; Court et al., 2003). On the other hand, UGT2B7*1c was recently associated with increased UGT2B7 protein levels and activity in human livers as well as higher AZT clearance in human subjects (Kwara et al., 2009). We did not observe any significant association between AcMPAG formation and polymorphism in UGT2B7, suggesting that the observed differences between diabetic and nondiabetic livers are independent of UGT2B7 polymorphism. However, the relatively small number of samples analyzed in the current study limits our ability to discern any genotype effect except to exclude it as a cause of the differences in AcMPAG formation between diabetic and nondiabetic livers.
Of all the immunosuppressant drugs currently available, MPA is the only compound that has been thoroughly investigated with regards to possible mechanism of action for causing GI toxicities. It has been reported that the effect of MPA-associated GI toxicity appears to be independent of the route of administration (Pescovitz et al., 2000). Moreover, it has been suggested that this toxicity may be caused by local GI concentration of AcMPAG rather than MPA or MPAG (Wieland et al., 2000; Picard et al., 2005). However, the exact mechanism is still unknown. Several possible mechanisms have been proposed, such as changes in purine synthesis and replication of epithelial cells as a result of inhibition of inosine monophosphate dehydrogenase activity (Shipkova et al., 2003). However, epithelial cells in the GI tract may not be wholly dependent on de novo purine synthesis and may be permeable to purines that are released into the intestine during digestion (Wilson and Wilson, 1962). In addition, formation of adducts with plasma proteins can directly interfere with cell function or trigger the immune system, leading to hypersensitivity and autoimmune reaction (Wieland et al., 2000). Up-regulation has been described for polymeric Ig receptor, glutathione-catalase, and CCAAT/enhanced-binding protein (Shipkova et al., 2004); and decreases in glucuronidation activity leading to lower AcMPAG concentration resulted in fewer GI toxicity (Yang et al., 2009).
Diabetes is associated with a decreased prevalence of MPA-associated GI toxicity when MPA is prescribed as an enteric-coated formulation of mycophenolate sodium versus the immediate released formulation of mycophenolate mofetil (result of MyGain study, unpublished communication with Novartis, Basel, Switzerland). Therefore, we speculate that down-regulation of UGT2B7 may decrease the formation of AcMPAG and thereby could decrease local irritation of the GI tract lumen and result in a lower incidence of GI toxicity in diabetic patients.
The present study has several limitations including the ex vivo nature of the observations, small sample size, and the question about quality of commercially obtained liver and kidney samples. To confirm the observation on UGT2B7 down-regulation by diabetes, a clinical pharmacokinetics study using a well known UGT2B7 substrate is warranted in diabetic patients closely matched with nondiabetic controls. Lack of tissue with known disease state such as diabetes is an important problem in characterizing ex vivo biotransformation in human. In general, most tissue banks in the United States lack extensive information about the medical history of the donor, disease state (i.e., diabetes status or type), or medications used by the donors. Hence, despite our best effort, only a small number of tissues with known diabetes status could be obtained. It will be important to verify our observations in a larger number of tissues from donors with a more comprehensive medical history (i.e., type of diabetes, presence of obesity, or insulin versus oral hypoglycemic agent therapy).
In conclusion, the findings of the present study provide evidence that diabetes significantly reduces the mRNA expression, protein level, and activity of human liver and kidney UGT2B7. In addition, the activity of UGT2B7 in the GI tract of diabetic patients would provide better insight about AcMPAG formation in the gut, but it was not studied in the current study. A major limitation of this study was the limited amount of information available regarding liver or kidney donors. Moreover, many questions remain unanswered concerning molecular mechanisms underlying the changes in UGT2B7 activity in diabetic patients.
Authorship Contributions
Participated in research design: Dostalek, Akhlaghi, and Court.
Conducted experiments: Dostalek, Hazarika, and Court.
Contributed new reagents or analytic tools: Akhlaghi.
Performed data analysis: Court, Hazarika, Dostalek, and Akhlaghi.
Wrote or contributed to the writing of the manuscript: Dostalek, Akhlaghi, Court, and Hazarika.
Other: Akhlaghi and Court acquired funding for the research.
Acknowledgments
We are grateful to Dr. Haiyan Xu (Division of Biology and Medicine, Brown University, Providence, RI) for the quantification of UGT2B7 protein levels.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM061834] (to M.H.C.); the American Heart Association [Grant 0855761D] (to F.A.); and an investigator initiated grant from Novartis Pharmaceuticals (to F.A.). The RI-INBRE core facility used in this study was funded by the National Institutes of Health National Center for Research Resources [Grant P20-RR016457].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding organizations.
The authors indicate no potential conflict of interest exists with any commercial entity whose products are described in the manuscript.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.036608.
-
ABBREVIATIONS:
- MPA
- mycophenolic acid
- MPAG
- pharmacologically inactive phenolic 7-O-glucuronide of MPA
- AcMPAG
- pharmacologically active acyl glucuronide of MPA
- GI
- gastrointestinal
- UGT
- uridine diphosphate-glucuronosyltransferase
- HEK
- human embryonic kidney
- AZT
- zidovudine
- HPLC
- high-performance liquid chromatography
- HLM
- human liver microsome
- HKM
- human kidney microsome
- PCR
- polymerase chain reaction
- TBARS
- thiobarbituric acid reactive substance.
- Received October 2, 2010.
- Accepted December 1, 2010.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}