


Abstract
Detailed cytochrome P450 (P450) inhibition profiles are now required for the registration of novel molecular entities. This method uses combined substrates (phenacetin, diclofenac, S-mephenytoin, bufuralol, and midazolam) with combined recombinant P450 enzymes (CYP1A2, 2C9, 2C19, 2D6, and 3A4) in an attempt to limit interactions with other more minor P450s and associated reductases. Kinetic analysis of single substrate with single P450 (sP450) yielded apparent Km values of 25, 2, 20, 9, and 3 μM, for CYP1A2, 2C9, 2C19, 2D6, and 3A4, respectively. Combined substrates with combined P450s (cP450) yielded apparent Km values of 65, 4, 19, 7, and 2 μM. Selectivity of the substrates for each P450 isoform was checked. Phenacetin proved to be the least selective substrate. However, the ratio of the various P450s was modified in the final assay such that metabolism of phenacetin by other enzymes was ∼20% of the metabolism by CYP1A2. IC50 determinations with α-naphthoflavone (0.04 μM), sulfaphenazole (0.26 μM), tranylcypromine (9 μM), quinidine (0.02 μM), and ketoconazole (0.01 μM) were similar for sP450 and cP450 enzymes. The assay was further evaluated with 11 literature compounds and 52 in-house new chemical entities, and the data compared with radiometric/fluorescent values. The overall protein level of the assay was reduced from the original starting point, as this led to some artificially high IC50 measurements when compared with existing lower protein assays (radiometric/fluorometric). This method offers high throughput P450 inhibition profiling with potential advantages over current radiometric or fluorometric methods.
There has been a considerable increase in publications in recent years regarding the use of mass spectrometry for assessing in vitro drug-drug interactions (Ayrton et al., 1998; Chu et al., 2000; Yin et al., 2000; Bu et al., 2001; Dierks et al., 2001; Zhang et al., 2001). This is partly due to technological advances in both instrument sensitivity and associated software.
Liquid chromatography/tandem mass spectrometry is the frontline choice for the majority of DMPK analyses (e.g., in vitro intrinsic clearance assays, Caco-2 cell permeability assays, protein binding, metabolite identification, and plasma analysis from in vivo studies). The use of high throughput methods in Lead Generation and Lead Optimization is now commonplace. The traditional methods for assessing the inhibition of P450s (liquid chromatography-UV, fluorometric, radiometric) can have limitations including cost of substrates, DMSO-sensitive assays (e.g., CYP3A4 fluorescence assays), interferences from NCEs (fluorescence/quenching), and safety considerations for radiometric assays. Mass spectrometry may therefore offer some advantages over these more traditional methods.
Many recent published MS P450 inhibition studies have focused on the use of human liver microsomes with “selective” substrates and (often) cocktail substrate incubations (Ayrton et al., 1998; Chu et al., 2000; Yin et al., 2000; Bu et al., 2001; Dierks et al., 2001; Zhang et al., 2001). However, it is estimated that ∼30% of human P450 isoforms remain uncharacterized (Shimada et al., 1994; Lewis, 2000) and true selective substrates for a single P450 isoform have proven to be elusive. Coupling this information with the low expression levels of certain major drug-metabolizing enzymes in HLMs e.g., CYP2C19 and CYP2D6, the work in this study has been performed with the major five human hepatic P450s expressed in Escherichia coli (CYP1A2, 2C9, 2C19, 2D6, and 3A4) (Rendic and Di Carlo, 1997), to minimize effects from other isoforms and to maximize turnover and thus improve signal/noise in the assay.
Advances in recombinant DNA technology and expression of the major human drug-metabolizing enzymes, with coexpressed NADPH reductase, in several cell-lines have made the use of recombinant P450s routine in many laboratories. Recent validation of E. coli expressed P450 enzymes as surrogates for their counterparts in HLMs (McGinnity et al., 1999) has provided further confidence in the use of these enzymes. The comparison of the kinetic properties of the E. coli expressed enzymes described here with human liver microsomes (HLMs) and appropriate substrates have been determined previously in this laboratory (McGinnity et al., 2000).
The method described here uses a cassette incubation of phenacetin (CYP1A2), diclofenac (CYP2C9), S-mephenytoin (CYP2C19), bufuralol (CYP2D6), and midazolam (CYP3A4), at their relative Km values with a cocktail of E. coli expressed P450s. In-house compounds were screened in the cP450 assay and the IC50 data were compared with predetermined radiometric (CYP2C9, 2C19, 2D6, 3A4) and fluorometric (CYP1A2) IC50 data. The compounds were chosen to represent a wide-range of physicochemical properties (variation in log D7.4, pKa and protein binding). The assay has been automated to provide IC50 data against these five P450s for up to 14 compounds at 6 concentrations per assay on a 96-well plate.
Materials and Methods
E. coli coexpressing the relevant P450s and human NADPH, P450 reductase, were purchased from Cypex (Dundee, UK). Previous studies have demonstrated that supplementation with cytochrome b5 is not required for these systems: kinetic parameters similar to both other recombinant systems and human liver microsomes for these five major isoforms have been reported (McGinnity et al., 1999, 2000).
S-mephenytoin, midazolam, bufuralol, 4′-hydroxymephenytoin, 1′-hydroxymidazolam, and hydroxybufuralol were purchased from Ultrafine Chemicals (Manchester, UK). Phenacetin, diclofenac, paracetamol, 4′-hydroxydiclofenac α-naphthoflavone, sulfaphenazole, tranylcypromine, quinidine, ketoconazole, all other cited chemicals and NADPH were purchased from Sigma Chemical (Poole, Dorset, UK). Develosil Combi-RP3 [3 μm, 35 × 3.0 mm i.d. column and guard cartridges C18 (ODS, octadecyl)] 4.0 × 2.0 mm were purchased from Phenomenex (Cheshire, UK).
Final Incubation Conditions. The final automated assay was based on an existing in-house 1A2 assay using a Tecan robot running Gemini/FACTS software. P450 concentrations were 5 pmol/ml (CYP3A4, 2C9, 2D6), 15 pmol/ml (CYP1A2), and 2.5 pmol/ml (CYP2C19) in 0.1 M potassium phosphate buffer, pH 7.4. All wells (controls, probe, and test inhibitors) contained DMSO at 1% v/v. The incubation volume was 200 μl with an assay stop time of 10 min via the addition of 200 μl of MeOH (final composition 1:1, aqueous/MeOH). Samples were shaken, chilled at –20°C for 2 h, spun at 3,500 rpm for 15 min, and the supernatant was transferred to vials for analysis.
Analytical Conditions. Mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in MeOH). Thirty microliters of sample were injected onto a Devosil C30 column. A fast gradient was developed. The gradient was as follows: 97% A (0–0.3min), 5% A (0.55–1.55 min), 97% A (1.6 min). The stop time was 2.5 min, the flow rate was 1.2 ml/min, and the column temperature was 40°C.
HPLC was performed with a Waters Alliance 2790 (Milford, MA) coupled to a triple quadrupole Quattro Ultima (Micromass, Manchester, UK) operating in ESI+ mode, with Masslynx 3.5 running in MRM mode (5 MRMs simultaneously, dwell time = 0.2 s) (Table 1).
MS conditions for the five metabolites used in all assays.
MS Ion Suppression Studies. MS ion suppression effects for electrospray ionization are well documented (Bonfiglio et al., 1999; Müller et al., 2002). All substrates are metabolized to between 1 and 5% (data not shown) during the 10 min incubation period. This large excess of substrate/metabolite ratio could lead to suppression of the detected metabolites. This is potentially complicated further by the effect of the rising concentrations of test inhibitors on the metabolite response. Depending on the test inhibitor profile, the metabolite areas could individually change by up to 100-fold or more, which may lead to differing ion suppression effects. To test for these potential interferences, each metabolite was incubated in the presence of all substrates and all other metabolites. Control metabolite was incubated alone without other substrates or metabolites, and the area measured was nominally assigned to 100%. The concentrations of metabolite were chosen to represent 20, 50, and 90% of control. All areas were then compared with the area obtained for control metabolite.
Time Linearity Assay (sP450). Individual incubations were carried out with a generic P450 concentration (25 pmol/ml) and at near Vmax conditions (∼5 times Km) based on values reported in the literature (Rodrigues et al., 1997; Dierks et al., 2001). Substrates were added to vials as methanol stocks and evaporated to dryness. Phosphate buffer (0.1 M, pH 7.4) was then added along with the calculated quantity of expressed P450 enzyme to form a total volume of enzyme/substrate mix of 10 ml. One hundred and seventy-eight microliters were added to wells on a 96-well microtiter plate (MTP) containing 2 μl of methanol (1% organic to represent later incubation conditions). All incubations were performed eight times simultaneously with an 8-channel pipette. The plate was preincubated for 10 min at 37°C in a warmed-air shaking incubator. The reactions were initiated by the addition of 20 μl of 10 mM NADPH, and the MTP plate was incubated at 37°C. The total volume of the incubations was 200 μl, and 15-μl aliquots were taken at 0, 2, 4, 8, 15, and 30 min. Aliquots were quenched by addition to an equal volume of ice-cold methanol. The samples were prepared and analyzed as described above. This assay design was used for all experiments described below unless otherwise stated.
Protein Linearity and Substrate Specificity Assay (sP450 and cP450). The conditions described above were repeated but with stop times of 10 min for all isoforms. The P450 concentrations used were 0, 6.25, 12.5, 25.0, 50, and 100 pmol/ml. For the substrate specificity assay, each substrate was incubated with each enzyme using the incubation conditions described above (10 min stop time, 25 pmol/ml P450, and at the reported Km).
Determination of Km and Vmax Parameters (sP450 and cP450). Incubations were carried out with ten substrate concentrations, encompassing the reported literature Km up to a value of five times Km. These were phenacetin (2.5–100 μM), diclofenac (1–25 μM), S-mephenytoin (5–200 μM), bufuralol (1–25 μM), and midazolam (0.5–12.5 μM).
The required volumes of methanolic substrate stocks were added to 7 ml of incubation vials and evaporated to dryness. The substrates were dissolved in a volume of buffer/enzyme solution to give a total volume of 5 ml. The sP450 assay used 25 pmol/ml of CYP3A4, 2C9, and 2D6; 75 pmol/ml of CYP1A2; and 12.5 pmol/ml of CYP2C19. The reactions were also carried out using a cP450 assay at 162.5 pmol/ml total P450 concentration (0.27 mg/ml). Experimental conditions were as above.
IC50 Determination of Probe Inhibitors (sP450 and cP450). All incubations were carried out at the measured Km values using the incubation conditions described above. IC50 determinations were determined (sP450) for standard inhibitors of the relevant P450s [α-naphthoflavone (CYP1A2), sulfaphenazole (CYP2C9), tranylcypromine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4)], and literature compounds were chosen to validate further the measured IC50 values (miconazole, 4-methylimidazole, mexilitine, lidocaine, omeprazole, dihydroergotamine, troleandomycin, nifedipine, ketoconazole, apigenin, haloperidol). The P450 concentrations were 25 pmol/ml (CYP3A4, 2C9, and 2D6), 75 pmol/ml (CYP1A2), and 12.5 pmol/ml (CYP2C19). A predilution plate was prepared from methanolic stocks of the individual inhibitors to give the concentrations 100× higher than required in the final assay. Two microliters of the inhibitor solutions were added to the substrate/enzyme mix (178 μl), and the plate was preincubated for 5 min at 37°C. The reaction was initiated via the addition of 20 μl of 10 mM NADPH in phosphate buffer. Incubations were carried out with ten inhibitor concentrations to span the known literature IC50 values. These were α-naphthoflavone (0.01–3 μM), sulfaphenazole (0.01–3 μM), tranylcypromine (0.1–30 μM), quinidine (0.001–0.3 μM), and ketoconazole (0.01–3 μM). The 11 literature compounds were diluted to give final concentrations of 0.15 to 50 μM, to mimic the final working assay.
The cP450 reactions were performed in the same way, but a combined substrate and a combined inhibitor solution were used with CYP3A4, 2C9, and 2D6 (25 pmol/ml), CYP1A2 (75 pmol/ml), and CYP2C19 (12.5 pmol/ml), to give a total P450 concentration of 162.5 pmol/ml.
Automated IC50 Determination of Test Inhibitors (sP450 and cP450). The conditions described above for the IC50 determinations were used to estimate the IC50 values for a range of literature compounds (see above). The assay was performed on a Tecan robot and the MTP layout is shown in Fig. 1. This time, all wells contained DMSO at 1% final concentration, to simulate the final working assay. The probe inhibitors above were included in both assays.
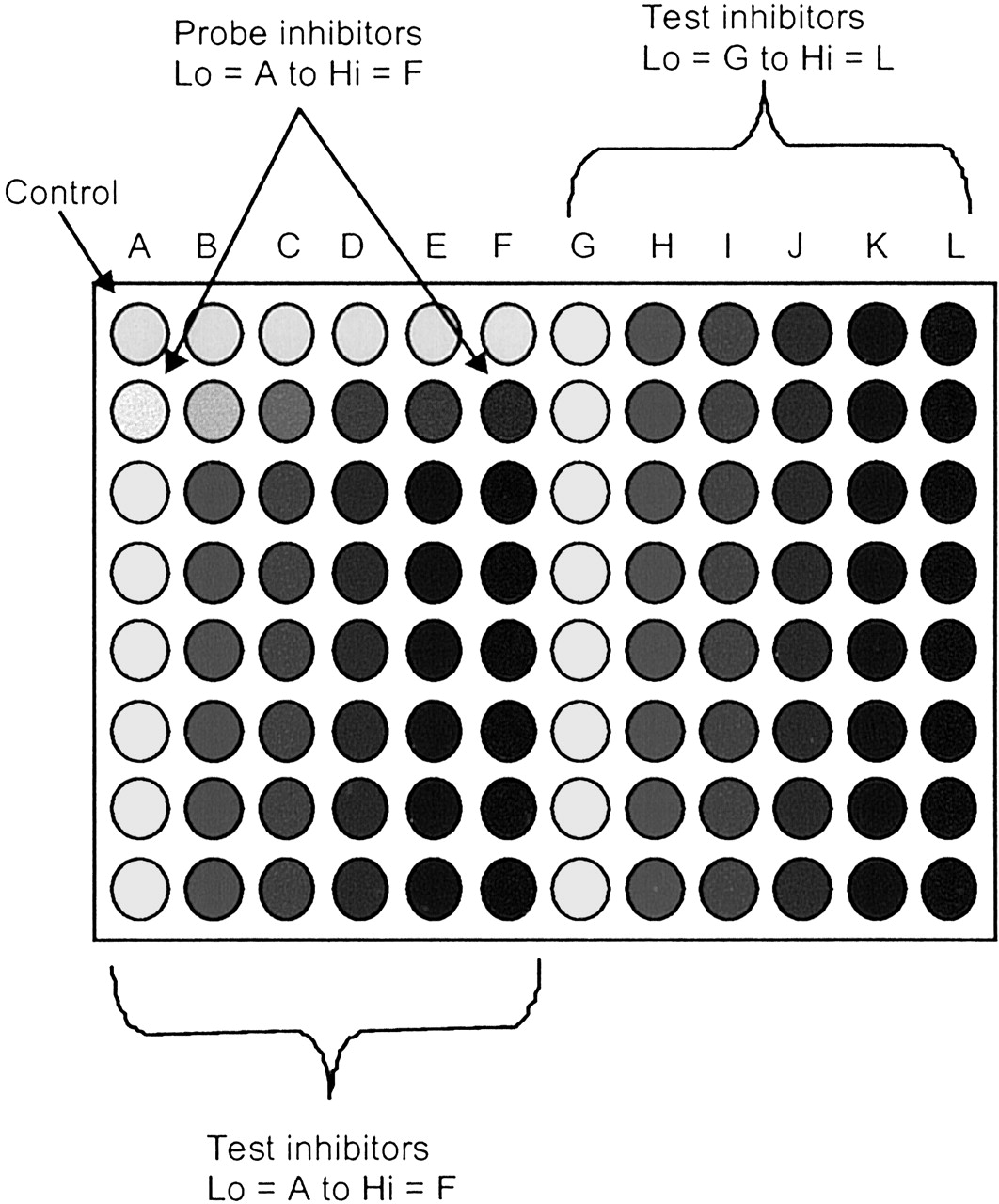
MTP layout for automated assay.
Final Automated IC50 Determination of Test (In-House NCEs) Inhibitors (cP450). The assay was identical to that described above. Two protein concentrations were used during method development along with the radiometric/fluorometric protein concentrations described in Table 2. The protein conditions used in the final automated assay are given in Table 2.
Protein concentrations used in all described assays.
Automated IC50 Determination of Probe/Test Inhibitors via Radiometric and Fluorometric Assays (sP450). All reference IC50 determinations were measured on the standard assays used in this laboratory, which are similar to those described by Moody et al. (1999). Enzyme sources and reagents were identical to the MS assays where possible.
Radiometric/fluorometric assay conditions: CYP2C9, 2C19, 2D6, and 3A4 were 14C-demethylation assays using naproxen, diazepam, dextromethorphan, and erythromycin with E. coli expressed P450s. For the CYP1A2 assay, ethoxyresorufin O-deethylation was used in an automated fluorescent-based assay with E. coli expressed CYP1A2 (Riley et al., 1995).
Data Analysis. Kinetic parameters were determined by linear or nonlinear regression using Microsoft Excel (Redmond, WA), Microcal Origin 6.0 (OriginLab Corporation, Northampton, MA), or WinNonLin 3.1 (Pharsight, Mountain View, CA). IC50 values were determined by linear transformation within Microsoft Excel.
Results
MS Ion Suppression Studies. Each metabolite was separated chromatographically on the chosen gradient (Fig. 2). They were also separated from the more lipophilic substrates, which tended to elute after all metabolites. Although substrate/metabolite ion suppression effects are unlikely because of this separation, an experiment was designed to probe this further. Each substrate was incubated in the presence of other substrates plus other metabolites (added at 20, 50, and 90%), and the areas measured were compared against a control metabolite (100%) in methanol/water only.
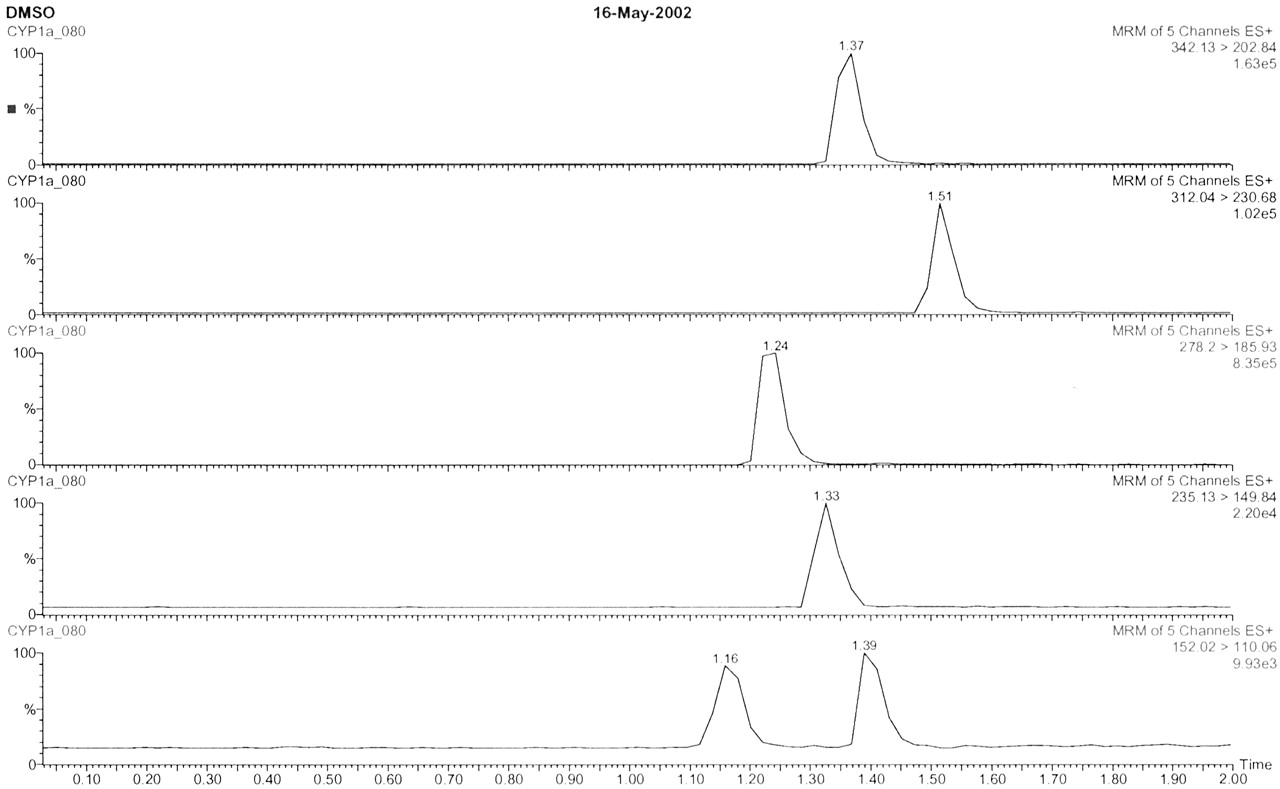
Control chromatograms of all metabolites.
The second peak at 1.39 min in the lower trace is due to the thermal degradation of phenacetin to paracetamol.
The measured mean thresholds and standard deviations were 21.7% (±1.9), 51% (±2.0), 90.3% (±1.7), and 99.5% (±1.4). Therefore, no significant ion suppression was observed for any of the metabolites in the presence of other substrates and metabolites.
Selection of MRM Method for 1′-Hydroxymidazolam. All metabolites were optimized for cone voltage and collision energy via loop injection. However, as midazolam is metabolized at two distinct points of the molecule by CYP3A4, a careful choice of MRM was taken. Both 1′-hydroxymidazolam and 4′-hydroxymidazolam lose water readily (342 > 324), and although this was a large fragment it was not used due to this nonspecificity. However, 1′-hydroxymidazolam gave a unique fragment (342 > 203), which was chosen for all quantification.
Time and Protein Linearity (sP450). Time linearity experiments were performed over 30 min. Reactions were linear up to 30 min for CYP1A2, 2C9, and 2C19. However, the formation of 1′-hydroxymidazolam was linear only to approximately 10 min. As the final assay was to be run with a cocktail of all five P450s, a stop time of 10 min was adopted for all remaining assays.
Protein linearity experiments were determined at six protein concentrations, spanning the probable final choice of concentration. CYP1A2, 2D6, and 2C19 were linear to 145, 100, and 100 pmol/ml, respectively. CYP3A4 and 2C9 were linear to approximately 50 and 40 pmol/ml (data not shown).
Substrate Specificity (sP450). Substrates were incubated at their assumed Km (Dierks et al., 2001; Lin et al., 2001) in the presence of each P450, to check for their specificity. Amount of quantified product formed was set at 100% for each prototypic substrate/enzyme pairs. Turnover by other P450s were then expressed as a percentage of this control rate. All five substrate/P450 combinations are shown graphically in Fig. 3.

Substrate specificity with individual enzymes phenacetin (A), diclofenac (B), S-mephenytoin (C), bufuralol (D), midazolam (E).
Selectivity profiles similar to those described by Dierks et al. (2000), for bufuralol, midazolam, S-mephenytoin, and diclofenac. S-mephenytoin oxidation was found to be specific for CYP2C19. Bufuralol oxidation was catalyzed by CYP2C19 (5%) and CYP3A4 (0.5%). Midazolam 1′-oxidation was catalyzed via CYP2C19 (2%), and diclofenac was metabolized to 4′-hydroxydiclofenac via CYP2C19 (3%) and CYP3A4 (2%). All were deemed acceptable for further work. Phenacetin proved to be a less selective substrate under the conditions chosen. CYP2C19 produced more paracetamol than CYP1A2 (157%) with contributions from CYP2D6 (26%), CYP3A4 (19%), and CYP2C9 (8%).
Determination of Km and Vmax Parameters (sP450 and cP450).Km and Vmax values were determined for individual substrates with individual enzymes. Final P450 concentration was 25 pmol/ml (CYP3A4, 2C9, 2D6), 75 pmol/ml (CYP1A2), and 12.5 pmol/ml (CYP2C19). Incubations were carried out with a range of substrate concentrations, spanning the reported literature Km, up to a value of 5 times Km.
Phenacetin, S-mephenytoin, and bufuralol obeyed classical Michaelis-Menten kinetics as expected. However midazolam and, to a certain extent, diclofenac exhibited nonclassical relationships. These data were fitted to a substrate inhibition model as described previously (Riley and Howbrook, 1998).
Km and Vmax values were then determined for cP450. Summarizing, the Km and Vmax values were similar between sP450 and cP450. The nonclassical Michaelis-Menten kinetics were exacerbated for midazolam and diclofenac. S-mephenytoin also exhibited depressed rates at high substrate concentrations, but this may have been due to poor solubility (which had been observed during method development), or other substrates competing for CYP2C19 at high substrate concentration. Representative plots for midazolam (nonclassical kinetics) and bufuralol (classical kinetics) are given in Fig. 4, and all results are summarized in Table 3.

Velocity/substrate plots for midazolam (A) and bufuralol (B) sP450 and cP450.
Measured kinetic parameters (sP450 and cP450).
IC50 Determination of Probe Inhibitors (sP450 and cP450). The IC50 values of the probe inhibitors α-napthoflavone (CYP1A2), sulfaphenazole (CYP2C9), tranylcypromine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4) were determined manually at the Km values described above with sP450 and cP450. The IC50 values for all isoforms were similar in the cP450 versus the sP450 assays. The values obtained in both assays were compared using a two-tailed paired t test and shown not to be significantly different (p = 0.32). IC50 plots are given in Fig. 5.

Representative IC50plots for CYP1A2 (A), CYP2C9 (B), CYP2C19 (C), CYP2D6 (D), and CYP3A4 (E)—sP450 and cP450.
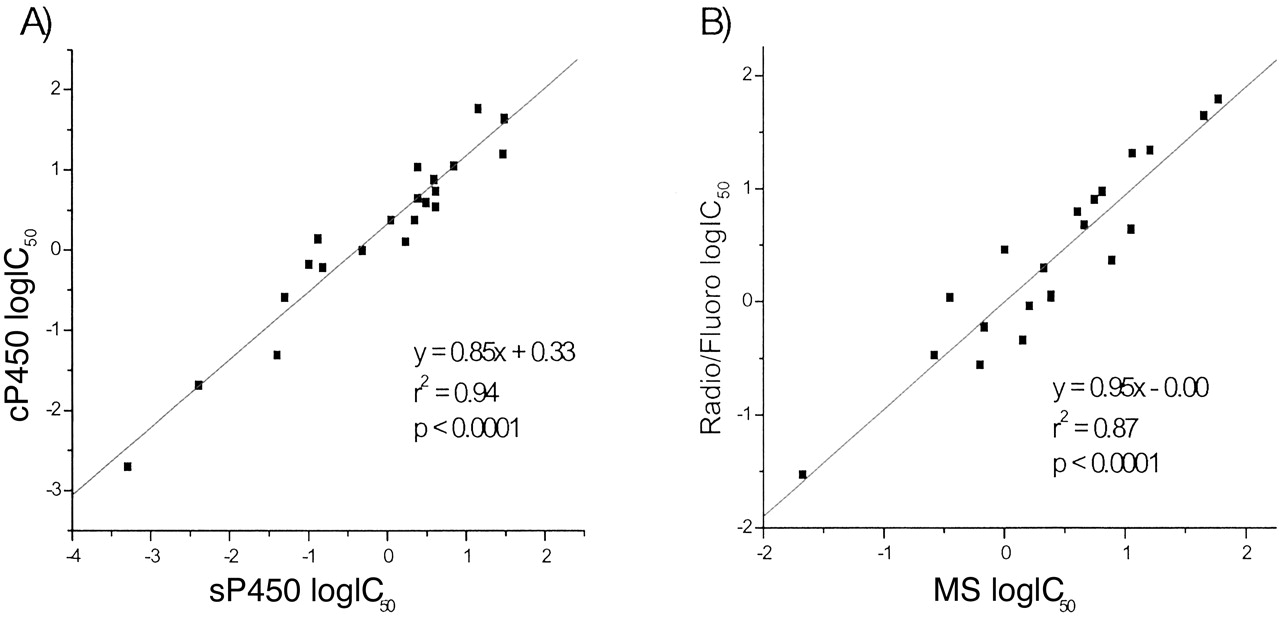
Automated IC50 Determination of Test Inhibitors (sP450 and cP450). IC50 determinations for 11 literature compounds were obtained from sP450 and cP450 and compared (miconazole, 4-methylimidazole, mexilitine, lidocaine, omeprazole, dihydroergotamine, troleandomycin, nifedipine, ketoconazole, apigenin, haloperidol). Generally, a good correlation (r2 = 0.94, p < 0.0001) was observed between both assays, with an overall higher IC50 shift for cP450 (2-fold, from y-axis intercept). The IC50 data for all isoforms was plotted on a single graph (Fig. 6). The cP450 data were then plotted against in-house radiometric (CYP2C9, 2C19, 3A4, and 2D6) and fluorometric (CYP1A2) determined data sP450. Again, a good correlation (r2 = 0.87, p < 0.0001) was observed between the sets of data (Fig. 6).

IC50comparisons between sP450 and cP450 (A) and cP450 MS and sP450 radiometric/fluorometric assays (B) with literature compounds (miconazole, 4-methylimidazole, mexilitine, lidocaine, omeprazole, dihydroergotamine, troleandomycin, nifedipine, ketoconazole, apigenin, and haloperidol).
Many data points could not be represented graphically as IC50 values >50 μM were observed for certain compound/isoform combinations in both sP450 and cP450.
Automated IC50 Determination of NCE Test Inhibitors (cP450). Fifty-two in-house NCEs were run in the cP450, and the IC50 values generated were compared with the traditional radiometric/fluorometric values generated in our lab with E. coli expressed P450s (sP450). Generally, good agreement was observed between assays (data not shown), but some chemical series gave higher IC50 values in the MS assay (e.g., worst-case dataset; Fig. 7). As evident from the similar gradients, intercepts and r2 values of the data in Fig. 7, this phenomenon was apparent for all P450s for all compounds. Following regression analysis, the MS IC50 values were shifted, on average, 10-fold higher compared with the radiometric value. In addition, the spread of IC50 data was limited in the MS method versus the radiometric method.

cP450 MS data versus sP450 radiometric data.
The data does not include CYP1A2 as all IC50 values were >100 μM.
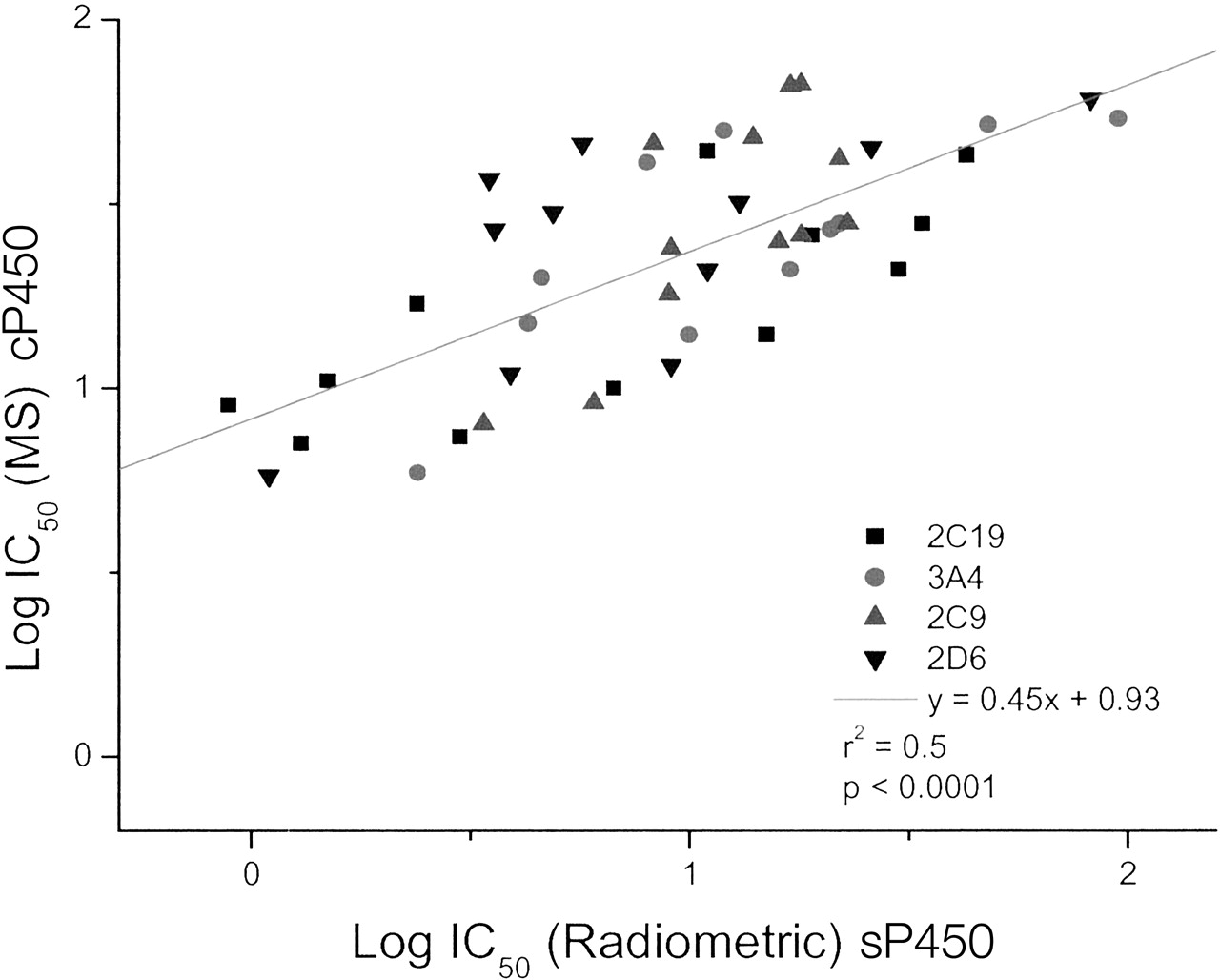
Finally, the cP450 experiment was repeated but with a 5-fold decrease in overall protein concentration (33 pmol/ml, 0.05 mg/ml). This total protein concentration resembles more closely those used in the traditional radiometric/fluorometric methods used in this laboratory (Table 2). These data were again plotted versus the traditional radiometric/fluorometric values generated with E. coli expressed P450s. The data are depicted graphically in Fig. 8.

cP450 MS data versus sP450 radiometric data at 1/5 total protein concentration.
The data does not include CYP1A2 as all IC50 values were >100 μM.
The line of best-fit gives an improved r2, intercept = 0, and the spread of IC50 data are now similar between both axes (compare Fig. 7). Furthermore, statistical analysis of the data for the high protein (Fig. 7) and low protein assays (Fig. 8) using a two-tailed paired t test confirmed them to be significantly different (p = 3.37E-10).
Discussion
This paper describes an MS-based cocktail P450 inhibition screen, using selective substrates for CYP1A2, 2C9, 2C19, 2D6, and 3A4. This work describes the evolution of an original fully automated assay (Moody et al., 1999) to a more streamlined MS assay. The assay was validated with literature compounds and evaluated more thoroughly with in-house NCEs. Finally, the assay was fully automated on a Tecan robot. The assay described here uses human hepatic P450s functionally coexpressed with human NADPH-P450 reductase in E. coli. This decision to use recombinant P450s over HLMs in a cocktail assay was supported by the following considerations: 1) E. coli expressed P450s have been shown to be faithful surrogates for HLMs in terms of substrate specificity (McGinnity et al., 1999), similar Michaelis-Menten kinetics (McGinnity et al., 1999, 2000), and enzyme-inhibitor interactions (Moody et al., 1999); 2) the five major P450s used in this assay metabolize ∼90% of prescribed drugs; and 3) true single P450-specific substrates will probably remain elusive. This method minimizes potential unwanted interactions between substrates and other P450 enzymes. In addition, the MS method offers excellent signal/noise with very low protein levels, cutting costs dramatically (e.g., cP450 concentration = 32.5 pmol/ml versus 170 pmol/ml in sequential or parallel CYP1A2, 2C9, 2C19, 2D6, and 3A4 radiometric/fluorometric assays).
The final assay run-time was limited by the 1′-hydroxymidazolam product formation, which was only linear to approximately 10 min. Gascon and Dayer (1991) described the kinetics of midazolam with HLMs and also found that formation of 1′-hydroxymidazolam and 4′-hydroxymidazolam was linear up to 15 min only with HLMs. Product inhibition (1′-hydroxymidazolam and/or 4′-hydroxymidazolam formation) after 10 min could account for the nonlinearity.
As all P450s gave linear product formation with increasing protein concentration to at least 40 pmol/ml, an initial protein concentration of 25 pmol/ml was used. This concentration was adopted to give good signal/noise during the initial investigations. However, as the final assay was to be used as a cocktail P450 assay, careful use of total protein was considered to avoid nonspecific binding, which could possibly give an overestimation of the IC50 for test inhibitors (Obach, 1997; Austin et al., 2002).
MS ion suppression effects can particularly affect electrospray ionization (Bonfiglio et al., 1999, Müller et al., 2002). This effect can reduce analyte response due to competition for charge by coelution of endogenous material and more usually by the presence of large excess of excipient or other chemical (e.g., polyethylene glycol). As the substrates are present in the assay at their respective Km values (2–25 μM) and are only depleted by 1–4% over the time course, the possibility of MS ion suppression of detected metabolites exists. These studies demonstrated that ion suppression does not appear to be an issue. However, one cannot rule out the possible ion suppression of a test inhibitor, which may coelute with one of the measured metabolites. In a worse case scenario, this could lead to an underestimation of IC50 giving a false-positive.
Substrate specificity was checked for all substrates with each P450 at assumed Km values (literature values). Any promiscuity between enzymes for the substrates used in a cocktail assay could give false IC50 values in the final assay if the substrate was metabolized significantly by another P450. The crossover to other P450s with these substrates has been documented [diclofenac (Shen et al., 1999; Ngui et al., 2000), phenacetin (Kobayashi et al., 1999; Venkatakrishnan et al., 1999), bufuralol (Mankowski, 1999), diclofenac (Shen et al., 1999; Ngui et al., 2000)]. Similar profiles were observed for diclofenac, S-mephenytoin, bufuralol, and midazolam. Diclofenac is known to be a CYP3A4 substrate with a reported Km of 71 μM with recombinant CYP3A4 (Ngui et al., 2000). Similarly, Shen et al. (1999) suggested that CYP3A4 was responsible for a particular metabolic activation (which culminates in covalent binding) of diclofenac. CYP2C19-mediated 4′-hydroxylation was also observed in this lab as reported by Mancy et al. (1999), who measured a Km of 440 μM for 4′-hydroxydiclofenac with CYP2C19 expressed in yeast. Masimirembwa et al. (1999) also observed metabolism of diclofenac with CYP2C19 in a similar recombinant system.
As anticipated, S-mephenytoin 4′-hydroxylation was shown to be specific to CYP2C19 (Masimirembwa et al., 1999). Bufuralol was metabolized to a small extent by CYP2C19. Mankowski (1999) measured an apparent Km of 36 μM for the metabolism of bufuralol with baculovirus expressed CYP2C19. Midazolam 1′-hydoxylation was catalyzed by 3A4, with a very small contribution from CYP2C19.
All the above were deemed acceptably selective for a cocktail assay approach. In fact, the highest percent crossover was only 6% with CYP2C19-mediated metabolism of bufuralol. However, phenacetin was far less selective, with all five isoforms contributing to the metabolism to paracetamol. Turnover of phenacetin by CYP2C19-mediated metabolism was higher than CYP1A2 (157% relative to CYP1A2). Obviously, this was not workable for a cocktail assay. For phenacetin to be a valuable probe in the cocktail assay, the ratio of CYP1A2/CYP2C19 was modified. Therefore, it was decided to reduce the CYP2C19 concentration 2-fold to 12.5 pmol/ml and raise the CYP1A2 concentration 3-fold to 75 pmol/ml, to minimize contribution of CYP2C19-mediated phenacetin metabolism. The CYP1A2 concentration could be increased, as the product formation was linear to at least 100 pmol/ml protein concentration. The theoretical net effect of this would be to reduce CYP2C19 phenacetin metabolism to 26% of CYP1A2-mediated phenacetin metabolism. This was borne out experimentally (Fig. 9). In fact, this level was taken to below 20% as DMSO, which was used at 1% in the final assay, reduced CYP2C19 activity more than CYP1A2 (data not shown). Venkatakrishnan et al. (1999) reported that five other isoforms (CYP2A6, 2C9, 2C19, 2D6, and 2E1) metabolized phenacetin to varying extents, with CYP2C19 giving the lowest Km at 656 μM. Kobayashi et al. (1999) noted that CYP2E1 was responsible for the low affinity component of phenacetin de-ethylation.

Relative metabolism of phenacetin by CYP1A2 and CYP2C19 at varying protein concentrations.
More recently, Belle et al. (2000) also suggest that CYP2C19 is the high Km enzyme responsible for phenacetin dethylation in human liver microsomes, which supports data presented here.
In an experiment with all P450s and phenacetin only, the amount of paracetamol produced was 126% (relative to a single incubation of phenacetin with 75 pmol/ml CYP1A2, data not shown), again indicating that contribution of metabolism of phenacetin by other P450s was negligible at the final protein concentrations chosen (Table 1).
The kinetics of the sP450 and cP450 were determined at the new protein concentrations (Table 1). Any deviations in apparent Km/Vmax in the cP450 versus the sP450 could indicate selectivity issues/substrate and/or product inhibition etc.
Generally, the Km values agreed well between sP450 and cP450. However, the apparent Km for phenacetin metabolism in the cP450 was somewhat higher (65 μM) compared with the sP450 (25 μM). This is probably due to some contribution from the low affinity component of phenacetin de-ethylation by CYP2C9/2C19/2D6 (Fig. 2)
Midazolam and diclofenac exhibited nonclassical Michaelis-Menten kinetics in both the sP450 and the cP450 assays. Substrate inhibition has been previously reported for midazolam (Kronbach et al., 1989; Gorski et al., 1994; Perloff et al., 2000) and triazolam (Schrag and Weinkers, 2001). At low concentrations, hyperbolic relationship exists between substrate concentration and velocity, but at high concentrations of substrate, the rate decreases rather than reaching a plateau. Schrag and Wienkers (2000) investigated the kinetics of triazolam with HLMs and proposed a two-site binding model for triazolam resulting in either 1′-hydroxytriazolam or 4′ hydroxytriazolam. Substrate inhibition then arises via competition for reactive oxygen.
Overall, Km and Vmax values generated in our lab agreed well with the literature values against HLMs and recombinant P450s (Table 4). As the final assay was run as a mixture of recombinant P450s, one must consider possible interactions of various P450 isoforms with one another (Tan et al. 1997; Yamazaki et al., 1997; Backes et al., 1998). The E. Coli expressed P450s described here give similar Michaelis-Menten kinetics when compared with human liver microsomes (McGinnity et al., 1999). Moreover, the similar kinetic parameters obtained between sP450 and cP450 would suggest minimal effects of such competition between P450 isoforms and P450 reductase.
Measured vs. literature Km and Vmax values.
The IC50 values of the probe inhibitors α-napthoflavone (CYP1A2), sulfaphenazole (CYP2C9), tranylcypromine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4) against sP450, agreed well with literature values (Table 5) and were comparable with the values obtained in the cP450 assay.
Measured vs. literature IC50 values.
Fifty-two in-house NCEs were screened to further evaluate the assay. The physicochemical properties were typical of the majority of compounds screened during a lead identification or lead optimization project. Therefore, the compounds chosen had a log D7.4 range from 0 to 5.5; human plasma protein binding range of 40 to 99.5%; molecular weight range from 200 to 600; and comprised of a mixture of acids, bases, and neutrals. All the compounds had been tested previously in P450 inhibition screens (radiometric/fluorometric), giving a large dynamic range of measured IC50 values (1 to 100 μM). None of the compounds had obvious structural alerts (e.g., unsubstituted pyridine or imidazole) or potential reactive functional groups. The reason for the higher IC50 values for some of the 52 NCEs tested in the cP450 versus radiometric method was believed to be due to higher protein levels in the cP450 experiment. The compounds that were most affected were from one particular chemical series. These compounds were more lipophilic (log D7.4 >4) and had higher plasma protein binding (>90%) compared with other compounds.
When the MS assay was repeated using 1/5 lower protein concentration to mimic the standard radiometric assay (0.05 mg/ml), the IC50 values for the NCEs were in good agreement between both assays (Fig. 8). Moreover, the y intercept on the log radiometric IC50 versus log MS IC50 plots was close to zero for all the individual P450 isoforms, indicating similar trends for all isoforms.
In conclusion, the assay described offers a robust, high-throughput screen for determining IC50 values against CYP1A2, 2C9, 2C19, 2D6, and 3A4 using recombinant P450s. This fully automated method was validated with literature compounds and in-house NCEs.
Footnotes
-
↵1 Abbreviations used are: DMSO, dimethyl sulfoxide; NCE, new chemical entity; P450, cytochrome P450; HLM, human liver microsome; MTP, microtiter plate; cP450, combined substrates combined P450s; MS, mass spectrometry; MRM, multiple reaction monitoring; sP450, single substrate single P450.
- Received November 25, 2002.
- Accepted March 24, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}