


Abstract
The human mass balance study is the definitive study for the assessment of absorption, distribution, metabolism, and excretion (ADME) properties of a new chemical entity in humans. Traditionally this has been carried out by the administration of radiolabeled drug substances, typically 14C or occasionally 3H, as detection methods for these isotopes allow the absolute quantification of drug-related material (DRM) in blood, plasma, and excreta. Coupled with the use of analytical techniques such as liquid chromatography-mass spectrometry, a picture of the metabolic fate of a compound can be elucidated. In this study, we demonstrate the capabilities of 19F nuclear magnetic resonance (NMR) spectroscopy, applied as an alternative to radiolabeling, for the determination of mass balance and for metabolite profiling of an orally administered fluorinated drug. To demonstrate the capabilities of NMR, the study was conducted on remaining samples from a 14C human mass balance study conducted on Alpelisib (BYL719), a compound in late stage development at Novartis for the treatment of solid tumors. Quantitative 14C data were used to cross-validate the data obtained by NMR. The data show that, using 19F NMR, comparable data can be obtained for key human ADME endpoints including mass balance, total DRM determination in plasma and metabolite profiling and identification in plasma and excreta. Potential scenarios where NMR could be employed as an alternative to radiolabeling for the conduct of an early human ADME study are discussed.
Introduction
Human absorption, distribution, metabolism, and excretion (ADME) studies are a standard part of the development process for new drugs. In these studies, healthy volunteers or patients are administered the drug containing radiolabel, typically 14C, after which they are hospitalized for a time. During this period, blood (and plasma) samples are taken and excreta are completely collected. The objectives and design aspects of these studies have been extensively reviewed and are not discussed further here (Beumer et al., 2006; Roffey et al., 2007; Nijenhuis et al., 2016). The timing of the human ADME varies (Penner et al., 2012), but general consensus is that the study is done once proof of concept is declared and/or preferably once the phase II dose is known. Ideally, the results should be available before the start of phase III dosing (U.S. Food and Drug Administration, 2008). Having human ADME data during the initial phase 1 studies would be highly advantageous and would allow for streamlining of later activities. Such an approach was proposed by Obach et al. (2012). With this strategy, the human ADME (and supporting dosimetry studies) would be the first radiolabeled studies conducted in drug development. After this, the exposures of major circulating metabolites were then compared with toxicology species using “cold” methodologies, with the result that preclinical radiolabeled studies would only be needed in certain circumstances. Despite the attractiveness of the approach, it appears it has not been widely adopted in industry. Possible reasons for this could be the requirements for good manufacturing practice synthesis of drug products for human use (U.S. Food and Drug Administration, 2008) and the resources needed to prepare for and run a human absorption, distribution, metabolism, and excretion study (hADME). These studies are costly and time consuming, and there are clear difficulties in convincing company management to move costs to phase 1 of development when compound attrition rates are still high. Instead, the industry has preferred to use “cold” mass spectrometry-based approaches to investigate drug metabolites in plasma or blood left over from phase 1 clinical studies and to establish exposure coverage with toxicology species (Yu et al., 2007; Yi et al., 2010; Ma et al., 2010; Gao et al., 2010). These, or similar, procedures are used extensively across the industry since publication of the FDA MIST guidance (U.S. Food and Drug Administration, 2016). The main issue with mass spectrometry-based screening approaches is that, to provide quantitative data, standards of the compounds of interest are needed [it is important to note that for establishing human:animal metabolite exposure ratios, as presented by Gao et al. (2010), metabolite standards are not required]. Leftover radiolabeled samples from early preclinical studies can be used for this purpose, as can chemical or biologic synthesis of relevant metabolites after initial screening of the human samples. However, these have several drawbacks. Radiolabeled samples from preclinical studies may not contain the relevant metabolites, and in any case the trend in industry has been to push these studies as late as possible in development after phase 1 clinical studies are completed. Metabolite synthesis is also often time consuming and costly. Finally, although these approaches address circulating metabolites, they provide little insight into the likely major elimination pathways of the drug.
Another approach that has been used to quantify metabolites in human plasma, and later urine, is 1H NMR (Dear et al., 2008; Nedderman et al., 2011). The quantitative power of NMR has been known for several decades but was not used much in drug metabolism studies due to sensitivity limitations. Instrument costs and the need for highly skilled operators were likely also a factor. This has been, in part, addressed by the advent of high-field magnets and cryoprobes. 19F NMR has also been used for quantitative metabolite profiling in rat urine and bile (Lenz et al., 2002) and, more recently, to assess mass balance and drug metabolism in preclinical ADME studies (Mutlib et al., 2012; Hu et al., 2017). Fluorine is incorporated frequently into drugs to alter chemical properties, disposition, and biologic activity (Park et al., 2001) and is compatible with NMR quantitation because of its favorable intrinsic NMR properties including the 100% natural abundance and low background interference.
In this article, we used samples remaining from a human ADME study conducted with Alpelisib (BYL719) (James et al., 2015) to demonstrate the utility of 19F NMR for the determination of key hADME deliverables, such as mass balance, quantitative metabolite profiling in plasma and excreta, and determination of total DRM pharmacokinetics (PK) in plasma. The presence of 14C label in the samples was used to cross-validate the 19F NMR results.
Materials and Methods
Chemicals and Standards.
[14C]BYL719 was synthesized by the Isotope laboratory of Novartis, Basel, Switzerland (Supplemental Material) and was blended with unlabeled BYL719 (Furet et al., 2013) to achieve a specific radioactivity of 6.94 kBq/mg (0.188 µCi/mg). Principal metabolite M4 was supplied by Novartis (Supplemental Material). The chemical structures/formula of 19F NMR internal standards 1 [Fevipiprant, supplied by Novartis (Bala et al., 2005; Sykes et al., 2016)] and 2 (obtained from commercial sources) are displayed in Table 1. All other chemicals and solvents were of the highest analytical grade available and were obtained from commercial sources.
Details of internal standards used for 19F NMR quantification
Human ADME Study with BYL719.
Plasma, urine, and feces samples used for the 19F NMR investigations were obtained from an open label, oral dose ADME study where BYL719 was administered to four healthy male subjects. All subjects provided written informed consent before enrollment. The study followed the ethical principles of the Declaration of Helsinki and the ICH Harmonized Tripartite Guidelines for Good Clinical Practice and applicable local regulations (European Directive 2001/20/EC and US Code of Federal Regulations Title 21) and was approved by an independent ethics committee before site initiation. The clinical part of the study was conducted at PRA Intl., Zuidlaren, The Netherlands. Each subject received a single oral dose of 400 mg [14C]BYL719 in two gelatin capsules of 200 mg [14C]BYL719 each (total dose: 2.78 Megabecquerel (MBq), 75 µCi). Subjects were hospitalized for up to 8 days, during which time blood, plasma, urine, and feces were collected. Total radioactivity determination in blood, plasma, urine samples, and feces homogenate were made by liquid scintillation counting (LSC) measurements conducted in the bioanalytical laboratory of the study site. Further details of this clinical study and results are published elsewhere (James et al., 2015).
Radioactivity Determination in Excreta.
Radioactivity contents in urine and feces homogenates were determined at the bioanalytical laboratory of PRA Intl. These data were used for the comparison with 19F NMR data for the determination of mass balance. For urine, duplicate (1000 µl) aliquots were placed into 7 ml glass vials (Perkin Elmer, Waltham, MA), after which 5 ml of scintillation cocktail (Ultima Gold, Perkin Elmer) was added. After being vortex mixed for at least 5 seconds, each sample was placed in a Tri-Carb 3100 TR liquid scintillation analyzer at least 30 minutes before counting. The total 14C radioactivity of the samples was determined by counting until a statistical error (2-sigma) of 0.5% was obtained with a counting time of 10 minutes.
For feces homogenates, quadruplicate, accurately weighed (500 mg) aliquots were dried in a stove at 50°C for at least 3 hours. After the addition of 100 µl combustaid (Perkin Elmer) to the dry homogenates, the samples were combusted in a sample oxidizer model 307 (Perkin Elmer). Seven milliliters CarboSorb-E (Perkin Elmer) was used as an absorber agent for carbon dioxide. At the end of the combustion cycle, the absorber was mixed with 13 ml of the scintillant PermaFluor E. The samples were placed in the liquid scintillation analyzer for at least 60 minutes before counting. The total 14C radioactivity of the samples was determined by counting until a statistical error (2s) of 0.5% was obtained with a counting time of 10 minutes.
Radioactivity Determination in Plasma and Plasma Extracts.
Aliquots of plasma were solubilized in a mixture of Soluene 350 (Packard)/isopropanol (2:1 v/v). After complete dissolution, the samples were neutralized with hydrochloric acid (2 M) and mixed with Irgasafe-Plus liquid scintillation cocktail (Zinsser Analytic, Maidenhead, Berkshire, UK) for liquid scintillation counting. Aliquots of plasma extracts were measured directly in either 20 or 6 ml antistatic polyethylene vials (Packard BioScience, Groningen, The Netherlands) containing 5 ml Irgasafe-Plus liquid scintillation cocktail. All samples were assayed for 14C radioactivity in a LSC counter model Tri-Carb 2200CA using an external standard ratio method for quench correction.
Preparation of Calibration Samples for Quantitative 19F NMR Analysis.
For the quantification of total DRM in plasma and excreta by 19F NMR and for quantitative metabolite profiling by 19F NMR, calibration curves were prepared. A stock solution of BYL719 was prepared by solubilization of a weighed amount in CD3OD/D2O (7:3 v/v), containing either internal standard 1 or 2 at a set concentration. Calibration samples were subsequently prepared by serial dilution of this stock solution in CD3OD/D2O (7:3 v/v), containing either internal standard 1 or 2 at a set concentration.
Preparation of Individual Plasma Time Points for Analysis of Total DRM by 19F NMR.
From one subject, 3 ml of plasma was taken from time points 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 hours postadministration of the dose and each was placed into a separate 50 ml BD Falcon tube. The plasma aliquots were subsequently extracted as follows: to each sample, 6 ml of ice-cold acetonitrile was added, and the resulting suspension was vortex mixed for 30 seconds and subjected to ultrasound (ultrasonication) for 5 minutes in a water bath. This was followed by centrifugation (10 minutes, 8331 g; GS-15R centrifuge, Beckman), after which the supernatant was removed from the pellet. The pellet was resuspended by the addition of 1 ml of water, followed by vortex mixing (30 seconds) and ultrasonication (5 minutes). To this, 8 ml of ice-cold acetonitrile was added and the mixture was further vortex mixed (30 seconds) and subjected to ultrasonication (5 minutes). Finally, the mixture was centrifuged as described above, and the resulting supernatant was combined with the first. The resulting pellet was re-extracted a further two times, following the procedure described above. The final combined supernatant was evaporated under a stream of nitrogen at room temperature to a volume of approximately 1 ml and was transferred to a 2 ml polypropylene tube. The original 50 ml Falcon tube was rinsed twice with acetonitrile, and these rinses were combined with the rest of the extract. Finally, the extract was evaporated to dryness under nitrogen flow at room temperature.
Each extract was reconstituted by the addition of 700 µl CD3OD/D2O (7/3 v/v), which contained internal standard 1 at a concentration of 5 µg/ml. One hundred seventy microliters of each reconstitute was transferred to a 3 mm NMR tube and was submitted for analysis.
Preparation of Plasma Area Under the Curve Pool for Quantitative Metabolite Profiling.
For one subject, volumes of plasma from time points 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours postadministration of the dose were pooled to mimic an area under the curve (AUC) exposure, as previously described (Hamilton et al., 1981). The total pool volume was 8832 µl. The pool was split into eight polypropylene tubes (15 ml capacity), with each containing approximately 1.1 ml of plasma. The plasma samples were extracted as follows:
To each sample, 2.2 ml of ice-cold acetonitrile was added, and the resulting suspension was vortex mixed for 30 seconds and subjected to ultrasound (ultrasonication) for 5 minutes in a water bath. This was followed by centrifugation (10 minutes, 8331 g; GS-15R centrifuge, Beckman), after which the supernatant was removed from the pellet. The pellet was resuspended by the addition of 0.3 ml of water, followed by vortex mixing (30 seconds) and ultrasonication (5 minutes). To this, 2.2 ml of ice-cold acetonitrile was added and the mixture was further vortex mixed (30 seconds) and subjected to ultrasonication (5 minutes). Finally the mixture was centrifuged as described above, and the resulting supernatant was combined with the first. The resulting pellet was re-extracted a further two times, following the procedure described above. The supernatants were combined from all plasma aliquots, and the resulting solution was evaporated under a stream of nitrogen at room temperature to a volume of approximately 4 ml. This was transferred to a 10 ml glass tube. The original tube containing the combined supernatants was rinsed three times with water/acetonitrile (9/1 v/v), and these rinses were combined with the rest of the extract. After removal of an aliquot (400 µl) for LSC, the remainder was injected onto the prep–high-performance liquid chromatography (HPLC) system and fractionated as described below. Resulting dried fractions were reconstituted by the addition of 600 µl of CD3OD/D2O (7/3 v/v) that contained internal standard 2 at a concentration of 2 µg/ml, followed by vortex mixing and ultrasonication. The resulting solutions were transferred into 5 mm NMR tubes and were submitted for 19F NMR analysis
Preparation of Urine and Feces Samples for Analysis of Total DRM by 19F NMR.
From all subjects in the study, at time periods predose, 0–6 hours, 6–12 hours, 12–24 hours, 24–48 hours, 48–72 hours, and 72–96 hours, duplicate 2 ml aliquots of urine were weighed into 5 ml polypropylene tubes. In parallel, duplicate 0.4 ml aliquots were taken for determination of radioactivity by LSC. The 2 ml aliquots were dried under a stream of nitrogen at room temperature and were reconstituted by the addition of 1 ml CD3OD/D2O (7/3 v/v), which contained internal standard 1 at a concentration of 25 µg/ml. Reconstituted samples were vortex mixed (10 seconds), ultrasonicated (30 minutes), and vortex mixed again for a further 10 seconds. Two aliquots of 150 µl were taken for LSC analysis and 650 µl was transferred into 5 mm NMR tubes for 19F NMR analysis.
From all subjects in the study, at time periods predose, 0–24 hours, 24–48 hours, 48–72 hours, 72–96 hours, 96–120 hours, 120–144 hours, and 144–168 hours, about 0.2 ml aliquots (duplicate) of fecal homogenate were taken for determination of radioactivity by LSC. In parallel, triplicate 1 g aliquots of feces homogenate were weighed into 15 ml polypropylene tubes. The 1 g aliquots were extracted as follows: to each sample, 5 ml of ice-cold acetonitrile was added, and the resulting suspension was vortex mixed for 10 seconds and subjected to ultrasound (ultrasonication) for 30 minutes in a water bath. This was followed by centrifugation (10 minutes, 10,000 g; Allegra 64R centrifuge, Beckman Coulter), after which the supernatant was removed from the pellet. The pellet was re-extracted by the addition of 1 ml of acetonitrile, followed by vortex mixing, ultrasonication, and centrifugation as described above. After removal of the supernatant, the pellet was extracted a third time in an identical manner. Supernatants from each extraction step were combined and evaporated to dryness under nitrogen. Samples were reconstituted by the addition of 1 ml CD3OD/D2O (7/3 v/v), which contained internal standard 1 at a concentration of 25 µg/ml. Reconstituted samples were vortex mixed (10 seconds), ultrasonicated (30 minutes), and vortex mixed again for an additional 10 seconds. Two aliquots of 150 µl were taken for LSC analysis, and 650 µl was transferred into 5 mm NMR tubes for 19F NMR analysis.
Preparation of Urine and Feces Pools for Quantitative Metabolite Profiling.
Urine was pooled across the time range 0–144 hours by taking equal percentages of the amount excreted from each time period (Subject 1000_00010). After removal of duplicate aliquots (0.5 ml) for LSC, 8 ml of urine was directly injected onto the prep-HPLC system and fractionated as described below. Resulting dried fractions were reconstituted by the addition of 600 µl of CD3OD/D2O (7/3 v/v), which contained internal standard 2 at a concentration of 2 µg/ml, followed by vortex mixing and ultrasonication. The resulting solutions were transferred into 5 mm NMR tubes and were submitted for 19F NMR analysis.
Feces homogenate was pooled across the time range 0–216 hours by taking equal percentages of the amount excreted from each individual time period (Subject 1000_00010). After duplicate aliquots (∼0.2 g) for LSC were removed, a sample of ∼1 g was weighed into a 15 ml polypropylene tube. The sample was extracted as described previously for the analysis of total DRM by 19F NMR. After evaporation to dryness under N2, the extraction residue was reconstituted by the addition of 4 ml of water, followed by vortex mixing (2 minutes) and ultrasonication (15 minutes). This reconstitute was transferred to a Waters autopurification system injection tube, after which the original 15 ml polypropylene tube was rinsed twice by the addition of 2 ml of water. Rinses were combined with the original reconstitute. After removal of duplicate aliquots of 100 µl for LSC, the remaining reconstitute was directly injected onto the prep-HPLC system and fractionated as described below. Resulting dried fractions were reconstituted by the addition of 600 µl of CD3OD/D2O (7/3 v/v), which contained internal standard 2 at a concentration of 10 µg/ml, followed by vortex mixing and ultrasonication. The resulting solutions were transferred into 5 mm NMR tubes and were submitted for 19F NMR analysis.
Semipreparative HPLC Conditions for Fractionation of Plasma Extract, Urine, and Feces Extract for Quantitative Metabolite Profiling by 19F NMR.
Semipreparative HPLC with fraction collection for quantitative metabolite profiling was carried out using a Waters Autopurification System equipped with MassLynx and the FractionLynx application manager. The column used was a Waters Atlantis Prep T3 (10 × 150 mm, 5 µm particle size) at room temperature. Post column flow was directed to a Gilson FC204 fraction collector operated in a time-slice mode. Separation and fractionation of BYL719 metabolites was accomplished using 10 mM ammonium acetate adjusted to pH 5.0 with acetic acid as mobile phase A and acetonitrile as mobile phase B. Flow rate was 5 ml/min. The semipreparative HPLC gradient was as follows: initial conditions were 10% mobile phase B, which increased to 70% at 10 minutes postinjection. From there, mobile phase B was increased to 95% at 11 minutes, where it was held (isocratic) up to 15 minutes. Finally, mobile phase B was decreased to 10% at 15.5 minutes, after which the column was re-equilibrated for subsequent injections. Post column flow was fractionated at 12 seconds per fraction into a 96 deep-well plate (Nunc 278752 U96 deepwell 96-well × 2 ml assay collection and storage microplate without lid, round bottom wells, non-treated natural polypropylene). Individual fractions were subsequently prepared for 19F NMR analysis as described above.
NMR Instrument Parameters Used for the Measurement of Total 19F in Urine, Feces Extracts, and Semipreparative HPLC Fractions.
One-dimensional 19F spectra were acquired at a temperature of 300 K using a Bruker 600 MHz Avance III NMR spectrometer equipped with a 5 mm 1H/19F-13C/15N/D CryoProbe with a z-gradient system. A total of 128 scans was accumulated for each sample using a standard proton inverse-gated decoupling pulse sequence with a relaxation delay of 7 seconds. Complex points (32,768) covering 34090.9 Hz were recorded at a transmitter frequency offset of −75 ppm. Data were zero-filled to 65,536 complex points prior to Fourier transformation, and an exponential window function was applied with a line-broadening factor of 1.0 Hz. The spectra were manually phase and baseline corrected and referenced to either the internal standard Fevipiprant (–61.0 ppm) or 3,5-bis(trifluoromethyl)phenol (–64.0 ppm). Based on these parameters the total acquisition time was approximately 15 minutes per sample.
Liquid Chromatography Tandem Mass Spectrometry; Conditions for Identification of Drug-Related Components Observed following 19F-NMR Analysis of Prep-HPLC Fractions.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) of prep-HPLC fractions with detectable 19F NMR signals was carried out either using an Agilent model 1200 HPLC coupled with a Waters Synapt HDMS or a Waters ultraperformance liquid chromatography system coupled with a Waters Synapt-G2 HDMS. The column used in both cases was a Waters Atlantis dC18 (2.1 × 150 mm, 3 µm) equipped with a precolumn of the same type (2.1 × 10 mm), which was placed in a column oven at 40°C. Mobile phase A was 10 mM ammonium acetate (aq) adjusted to pH 5.0 with acetic acid. Mobile phase B was acetonitrile. Flow rate was 0.25 ml/min. The HPLC gradient was as follows: initial conditions were 30% mobile phase B, which increased to 70% at 10 minutes postinjection. From there, mobile phase B was increased to 100% at 11 minutes, where it was held (isocratic) up to 17 minutes. Finally, mobile phase B was decreased rapidly to 30%, after which the column was re-equilibrated for subsequent injections.
Assessment of Potential Matrix Effect on the Measurement of Total 19F in Plasma Extracts, Urine, and Feces Extracts.
To assess any potential matrix effect on the 19F NMR measurement, a series of samples were prepared in control human urine, feces extract, plasma extract, and NMR solvents (CD3OD/D2O 7/3 v/v). In the case of urine, two aliquots of urine were evaporated to dryness under N2. For feces, two ∼1 g aliquots were extracted as previously described. For plasma, two ∼3 g aliquots were extracted as previously described. The resulting residues were reconstituted in CD3OD/D2O (7/3 v/v). The reconstitution solvent contained sufficient BYL719 and internal standard 2 to achieve, for the first aliquots, a concentration of 100 and 25 µg/ml, respectively. The final concentrations in the second aliquots were 1 and 2.5 µg/ml, respectively. A control sample without matrix (70/30 v/v CD3OD/D2O) was also prepared at both sets of concentrations. The final solutions were placed into 5 mm NMR tubes and were analyzed as described above. Measurements were carried out in quintuplicate to assess both the effect of the matrix and NMR instrument precision at the two BYL719 concentrations.
Results
Excretion Balance.
The percentage of dose excreted in each collection interval for both urine and feces and comparing 19F NMR and 14C measurements is shown in Fig. 1. For urine, the percentage of dose excreted up to 96 hours postdose was 13.4 ± 3.83%, based on total 14C measurement, and 12.3 ± 2.47% when measured by 19F NMR. For feces, up to 168 hours postdose, 80.9 ± 3.24% of the dose was excreted (14C measurement) compared with 83.7 ± 3.95% when measured by 19F NMR. With both means of measurement, excretion of the administered dose could be classified as complete (>90%). Good agreement between 19F and 14C measurements was obtained for all collection intervals for both urine and feces.

Mean cumulative excretion of dose in urine and feces after a single oral dose of 400 mg [14C]BYL719 (Alpelisib) to four healthy volunteers, determined by liquid scintillation counting and 19F NMR.
Total DRM in Collected Plasma Samples.
The concentrations of total DRM in plasma extract from 0.5 to 12 hours determined by 19F NMR analysis and compared with total 14C measurements of the same plasma extract are detailed in Fig. 2. Forty-eight hours after dosing, total DRM was below the limit of quantification by both 19F NMR and 14C measurements. At 24 hours after dosing, concentrations were measureable by 14C but not by 19F NMR. In our conditions, the limit of quantification by 19F NMR was estimated at 500 ng eq/ml for BYL719. The limit of quantification (LOQ) for the plasma analysis by LSC, which is dependent on the specific activity of the administered drug and the validated ranges of the instrumentation, was approximately 75 ng eq/ml for this study. Some variability was observed between the 19F NMR and 14C measurements (with maximum 19% difference and 3 samples above 10%). As a final step, AUC0–12 hours was calculated based on NMR and 14C measurements. The results were comparable, showing a 9% difference.

Concentrations of total DRM in individual plasma time points from a single healthy volunteer after a single oral dose of 400 mg [14C]BYL719 (Alpelisib) determined by liquid scintillation counting and 19F NMR. Integration of 19F NMR signals was performed manually.
Metabolite Profiling in Urine.
19F NMR analysis of individual fractions generated from the analysis of the 0- to 144-hour urine pool allowed reconstruction of a chromatogram (Fig. 3). Three peaks were observed. Subsequent analysis by LC-MS/MS showed that the early eluting peak (P1) was comprised of coeluting metabolites M1 and M12; the middle peak (P2) metabolites M3, M4, and M9; and the last peak (P3) BYL719. Structures of all metabolites observed in this study are shown in Fig. 4. 19F mass balance measurements indicated that 12.6% of the dose was excreted in urine over this time interval. Therefore, in terms of percentage of dose, 2.31% was attributed to BYL719; 9.43% to M3, M4, and M9; and 0.87% to M1 and M12. This compared well to results obtained with conventional 14C radioprofiling and mass balance measurements, where BYL719 represented 2.04%; M3, M4. and M9 9.15%; and M1 and M12 1.15% of the dose. Notably, metabolites coeluted under the preparative HPLC conditions used to prepare samples for 19F NMR, but were resolved with the HPLC method used in the original human ADME. This is discussed below.
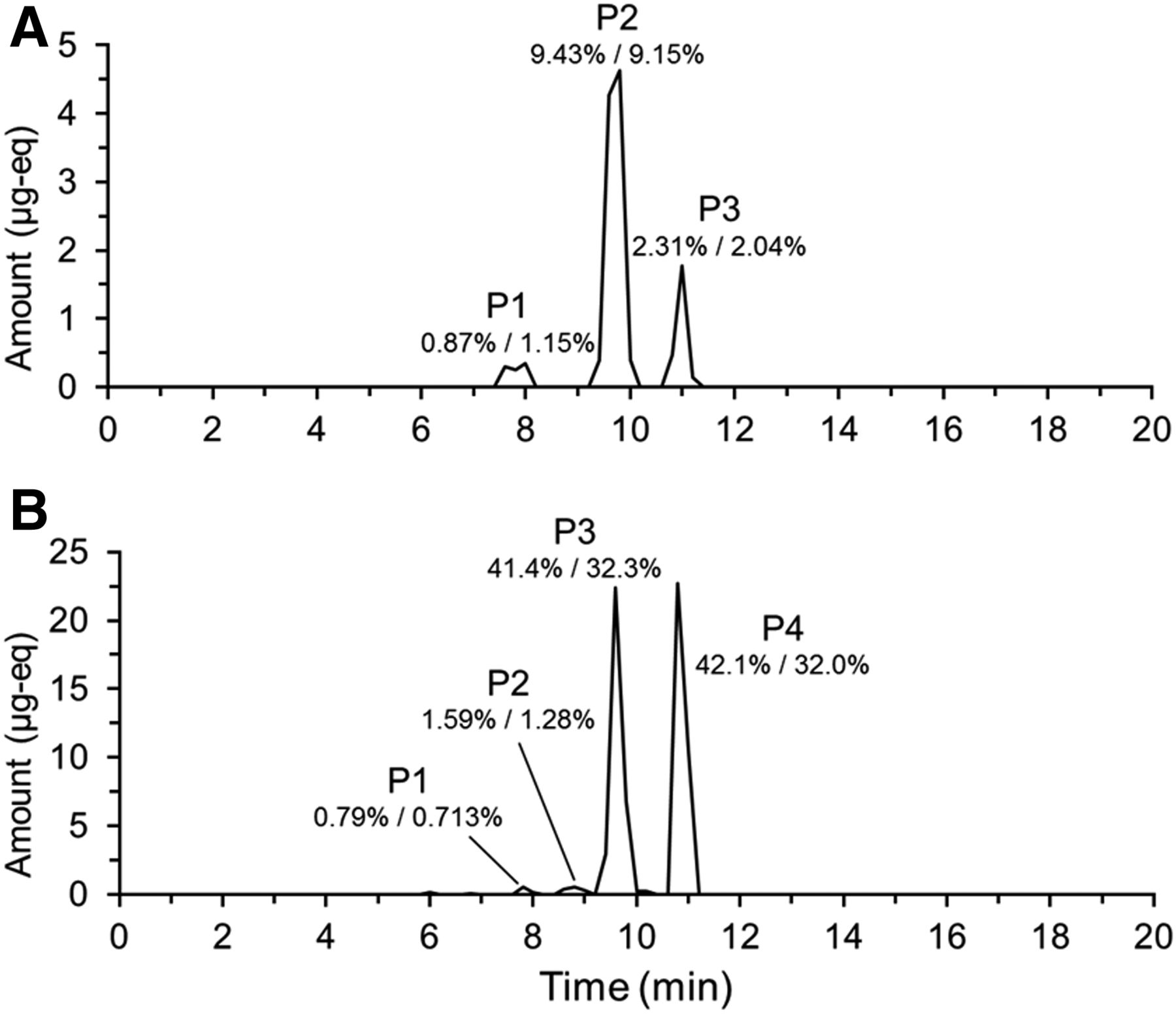
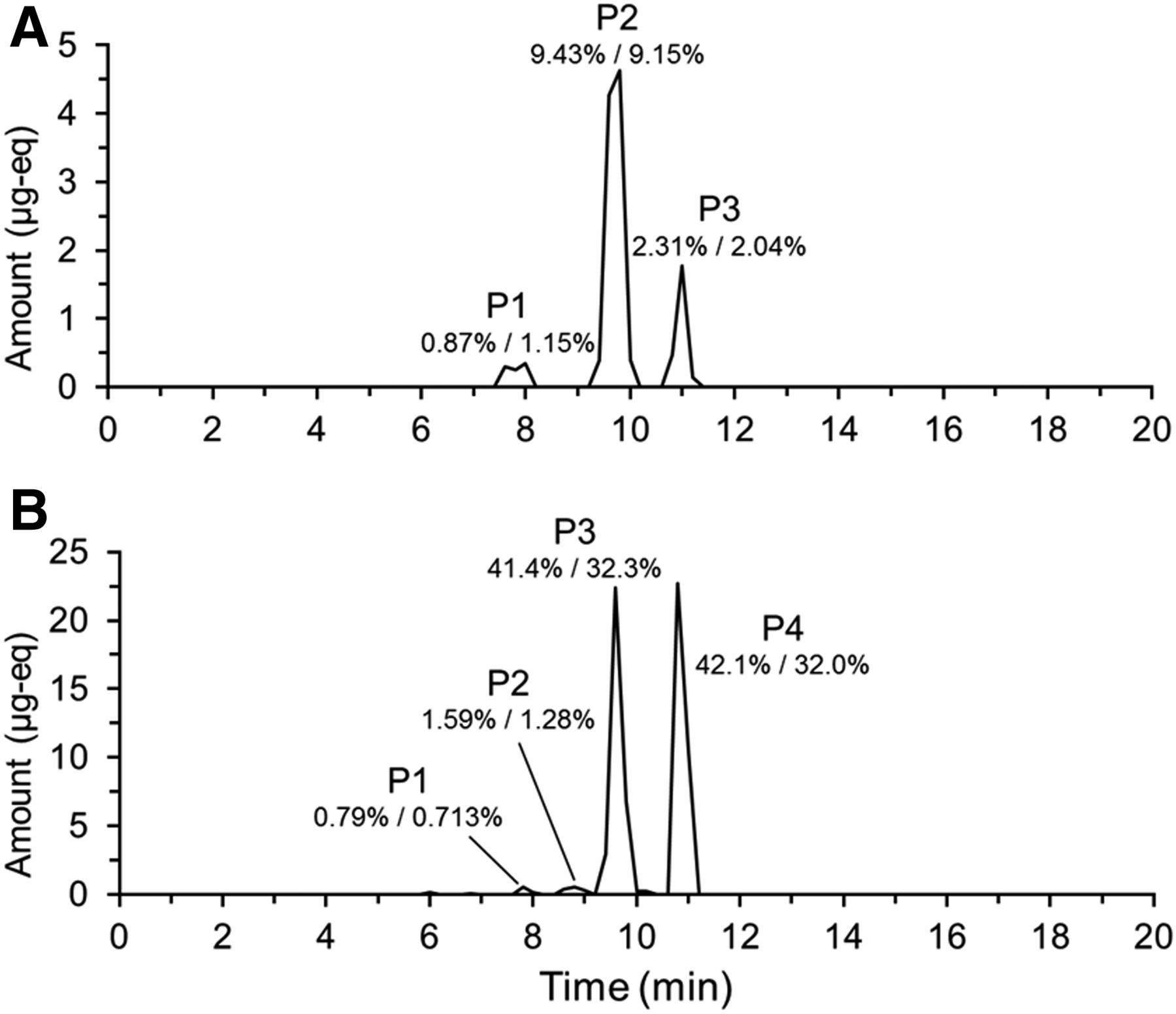
Reconstructed 19F NMR chromatogram showing the metabolic profile of BYL719 (Alpelisib) in (A; urine) and (B; feces). Chromatograms reconstructed from total 19F NMR quantification in individual fractions generated by time-slice fraction collection on a preparative-HPLC system. Metabolites were identified by LC-MS/MS analysis of fractions containing 19F. The percentages listed are the percentage of administered dose attributable to the components determined by 19F/14C.

Structures of metabolites of Alpelisib (BYL719) identified in human plasma, urine, and feces. The * indicates the position of the radiolabel.
Metabolite Profiling in Feces.
19F NMR of individual fractions generated from analysis of a 0- to 216-hour feces pool allowed reconstruction of a chromatogram (Fig. 3). Four peaks were observed (P1, P2, P3, and P4). Subsequent analysis by LC-MS/MS showed P1 consisted of metabolite M12; P2 consisted of coeluting M2 and M8; P3 consisted of coeluting M3, M4. and M9; and P4 was attributed to BYL719. 19F mass balance measurements indicated that 85.9% of the dose was excreted over this time interval. Therefore, in terms of percentage of dose, 0.79% was attributed to M12; 1.59% to M2 and M8; 41.4% to M3, M4, and M9; and 42.1% to BYL719. In the original human ADME, results from 14C radioprofiling were slightly different, with 0.713%, 1.28%, 32.3%, and 32.0% of the dose being attributable to the groups of metabolites, respectively. All metabolites detected in the human ADME, with the exception of a minor component M19 (0.5% of dose), were detected by 19F NMR.
Metabolite Profiling in Plasma.
19F NMR of individual fractions generated from analysis of a 0- to 24-hour pool allowed reconstruction of a chromatogram (Fig. 5). Two compounds were detected: M4, which represented 23.9%, and Alpelisib, which represented 76.1% of AUClast. 14C radioprofiling of individual plasma time points from 0 to 12 hours from the same subject showed M4 representing 28.8% and Alpelisib 63.7% of AUClast. Two minor metabolites, M3 and M12, which represented 0.13% and 1.33% of the 14C AUClast, respectively, were not detected with the 19F NMR method. By use of the total DRM in each plasma sample derived from 19F NMR measurements, an AUC0–12 hours of 28,869 nM⋅h was calculated. Considering the 19F chromatogram, this translates to an AUC0–12 hours of 6900 nM⋅h for metabolite M4 and 21,969 nM⋅h for BYL719. In the original human ADME, the derived AUC0–12 hours were 8990 nM⋅h and 19,900 nM⋅h for metabolite M4 and BYL719, respectively. It is important to note that in the case of the 14C-derived parameters, the presence of additional minor metabolites M3 and M12 and 14C lost during sample processing were taken into account in the calculation.

Reconstructed 19F NMR chromatogram showing the metabolic profile of BYL719 (Alpelisib) in a plasma AUC0- to 24-hour pool of a single subject. Chromatogram reconstructed from total 19F NMR quantification in individual fractions generated by time-slice fraction collection on a preparative-HPLC system. Metabolites were identified by LC-MS/MS analysis of fractions containing 19F. The percentages listed are the percentage of total observed DRM in the sample, determined by 19F/14C.
Assessment of Matrix Effect on NMR Measurements.
The NMR integrals (relative to internal standard) for BYL719 spiked into urine, feces extract, plasma extract, and NMR reconstitution solvent are shown in Fig. 6. The relative standard deviations are calculated from n = 5 measurements. The final concentrations of BYL719 in the NMR tubes were 100 and 1 µg/ml, respectively. From the data, it is clear that the matrix had negligible impact on the 19F NMR signal under our analytical conditions.

Chart showing the internal standard corrected 19F NMR integrals obtained from analysis of standard solutions of BYL719 (Alpelisib) and internal standard 2, spiked into urine, feces, plasma, and NMR solvents at analyte:internal standard concentrations of 100:25 and 1:2.5 µg/ml, respectively. Each sample was analyzed a total of five times by 19F NMR. Relative standard deviations from the five measurements are listed.
Discussion
NMR is/has been used for a number of applications in biomedical research and development, including metabolomics, in vivo magnetic resonance spectroscopy, and pharmaceutical analysis [drug identification, drug impurity characterization, degradation studies, drug isomeric composition, and analysis of counterfeits (Holzgrabe, 2010; Malet-Martino and Holzgrabe, 2011; Everett, 2015)]. Quantitative NMR has also been used for drug metabolism investigations, such as quantification of biologically isolated metabolites for use in pharmacological activity testing (Mutlib et al., 2011), as standards for quantitative assays to address MIST guidance concerns (Espina et al., 2009; Walker et al., 2011) or in preclinical in vivo efficacy models (Walker et al., 2014). It has also been used in preclinical in vivo studies to establish the urinary metabolic fate of 2-bromo-4-trifluoromethylaniline in rat (Scarfe et al., 1998,1999) and to conduct ADME in preclinical species (Mutlib et al., 2012). In human studies it has been applied to urinary mass balance and metabolite profiling (Skordi et al., 2004; Nedderman et al., 2011) and quantitative metabolite profiling in plasma (Dear et al., 2008). In these examples, either 1H or 19F NMR was used. 19F has advantages for the quantification of drugs and metabolites in complex biologic matrices, because of the extremely low level of endogenous fluorine-containing compounds (Martino et al., 2005). Fluorine is incorporated frequently into drugs to alter physicochemical properties (Park et al., 2001), which makes 19F NMR potentially applicable for the study of drug ADME.
In this study, we investigated whether 19F NMR could be used to achieve the following hADME objectives: 1) determination of mass balance, 2) determination of total DRM PK in plasma, 3) quantitative metabolite profiling in plasma (MIST guidance considerations), and 4) quantitative metabolite profiling in excreta to assign the major metabolic elimination pathways. Using leftover samples from the human ADME study with Alpelisib allowed for cross-validation of the NMR data with radioactivity data. Excellent agreement was obtained between 19F and 14C measurements for all of the listed objectives. Nevertheless, our experience conducting the study raised several points that require additional discussion.
NMR Quantitation.
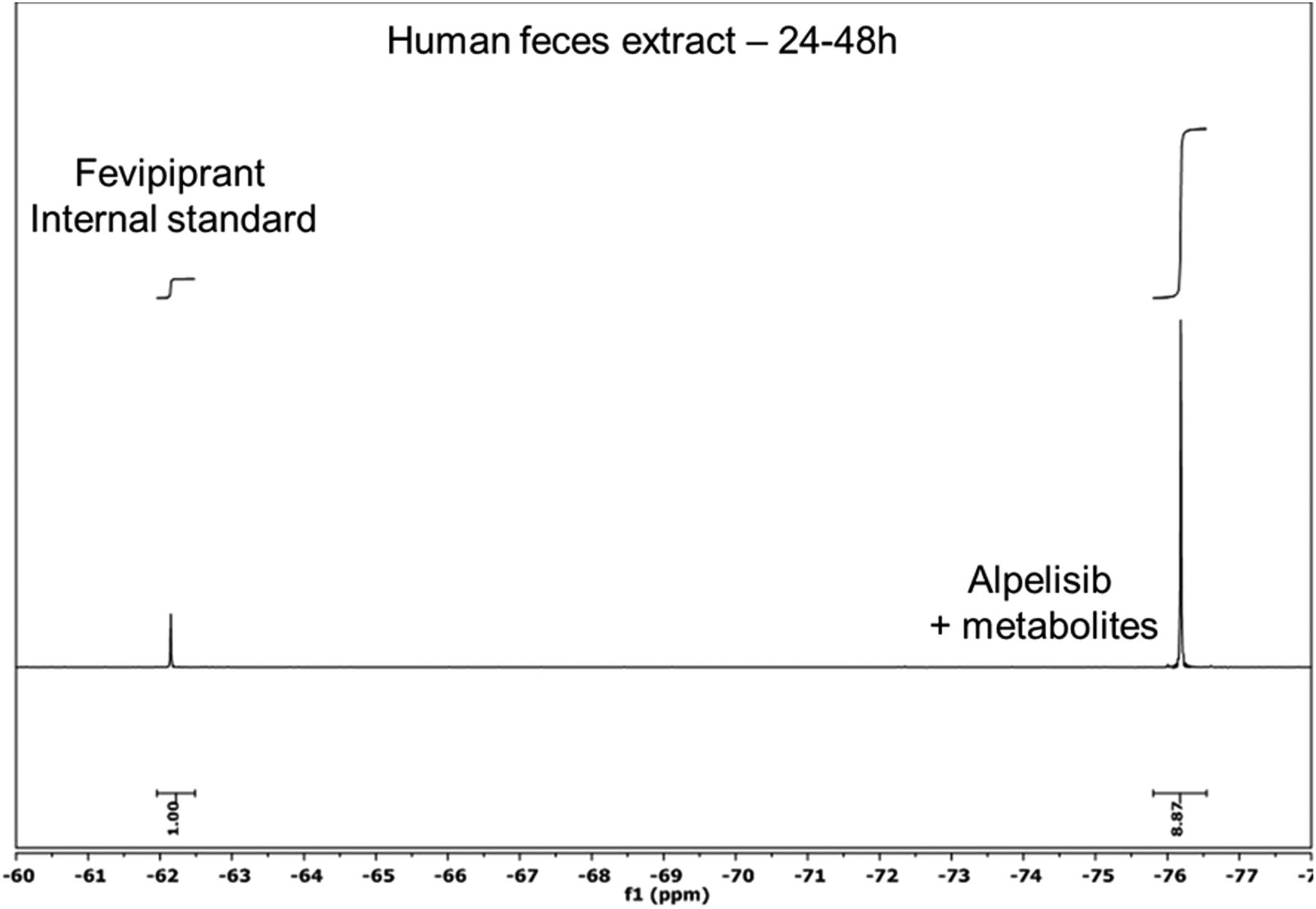
Quantification using NMR can be accomplished using internal or external standards (Holzgrabe, 2015). For quantification the internal reference method was chosen for this study, to assure the highest accuracy and precision (Cullen et al., 2013, Giraudeau et al., 2014). Key considerations that need to be taken into account when selecting a suitable internal standard are: 1) the fluorine signal does not overlap with those of the drug and metabolites, 2) the signal intensity should not differ more than 50-fold from the expected lowest and highest concentrations, and 3) to avoid off-resonance effects, the fluorine shift should not differ too much from the analytes of interest (Power et al., 2016). For fluorine NMR, use of an appropriate internal standard for a specific chemical shift region should not present problems, because a wide range of compounds could be used. In our study we selected either Fevipiprant or 3,5-bis(trifluoromethyl)phenol as internal standards, which were added to every sample. In Fig. 7, a typical one-dimensional 19F spectrum is shown, revealing the signals from the internal standard and Alpelisib + metabolites in human feces extract. Quantification is based on signal intensity as indicated in the figure. Although no evidence of matrix effects were observed in this study, it is known that matrix can have an impact on NMR quantitation, such as salt concentration. Although high salt concentrations result in “lossy samples” due to increased conductivity, this does not have any effect on relative signal integrals, because analytes and internal standards are equally affected (Voehler et al., 2006; Robosky et al., 2007). To minimize any additional matrix effects, calibration curves can be recorded with the standards being prepared in the matrix of interest. To avoid signal saturation effects, T1 relaxation times of the parent compound and the internal standard were determined and the relaxation delay adjusted accordingly.

Excerpt from typical one-dimensional 19F spectrum showing the signal of the internal standard Fevipiprant and of Alpelisib + metabolites. The spectrum was acquired on a 24- to 48-hour human feces extract. The major metabolites of Alpelisib were not resolved from the parent compound in the 19F NMR spectrum.
Mass Balance.
Measurement of mass balance relies on complete collection of excreta. If a 19F NMR approach is considered for a phase 1 study, collection of feces needs to be added to a cohort in the single ascending dose arm of the study. This is not commonly done, and key questions that need to be asked are 1) does the clinical unit selected have experience with feces collection and 2) how will the feces be processed? Internal experience suggests that it is difficult to convince teams to agree to feces collections for more than 5 days in a phase 1 study, despite limited additional cost, but it is critical for this approach so that it is possible to collect longer (minimum 7 days). Processing of stools can either be done at the clinical site, in the analytical laboratory of the sponsor company, or can be outsourced to a second contract resource organization familiar with feces homogenization procedures. From an analytical perspective, measurement of mass balance in urine is relatively simple, with minimal sample processing required. For feces, an organic solvent extraction needs to be performed to prepare samples for NMR analysis. Low extraction recovery, which cannot be measured using this approach, would contribute to low mass balance. The use of high-resolution magic angle spinning NMR may compensate for this, because feces homogenate could be loaded directly into the sample tube for measurement. High-resolution magic angle spinning NMR is a technology especially suitable for viscous or semisolid samples. It combines the advantages of sensitive high-resolution NMR probes with magic angle spinning technology for suppression of dipolar couplings known from solid-state NMR. It has successfully been applied to a wide range of inhomogeneous samples like gels, swollen polymers, resins, foodstuff, cells, and tissue samples (Power, 2011). However, the applicability for quantification of low molecular weight compounds in feces remains to be tested.
Total DRM PK.
In this study, total DRM in individual plasma time points was measured by 19F NMR. As for feces, plasma samples were extracted before analysis, which could lead to possible losses. A combination of lyophilization followed by high-resolution magic angle spinning NMR spectroscopy may account for this, as discussed above. However, this would likely only allow measurement of “free” compound-related material that was not extractable by protein precipitation. Covalently bound material, e.g., to plasma proteins, would probably not be quantifiable due to broadening of 19F resonances.
Metabolite Profiling.
We have demonstrated that 19F NMR can provide reliable quantitative metabolite profiles in plasma and excreta for the assessment of metabolite exposures and metabolic elimination pathways. As previously discussed, extraction recoveries cannot be measured for plasma and feces. This is the most likely explanation for the discrepancies that were noted in the 19F and 14C profiles for feces, because, in the latter case, column and extraction recoveries were taken into account in the calculation. A second limitation was coelution of metabolites in the excreta profiles. Development of chromatography methods that separate all metabolites of interest is more challenging with a preparative HPLC system. These limitations could be overcome by, for example, multiple repeated injections with fraction collection using a HPLC/ultraperformance liquid chromatography system. In addition, coelution may not be an issue for some compounds. Fluorine resonances are very sensitive to structural and steric changes, so it is feasible that a mixture of coeluting metabolites could be quantified individually (Hu et al., 2017). Finally, the fluorine group may be removed by metabolism. Although unlikely for a CF3 moiety, it is known that single fluorine atoms in a drug can be removed by oxidative defluorination (Park et al., 2001). This would make the metabolite unavailable for NMR quantification.
Overall Summary.
We have demonstrated that hADME objectives are achievable using 19F NMR. The availability of complementary 14C data demonstrates that the NMR data can be of high quality, despite the limitations discussed above. The main advantage of this approach is that it could be applied in a phase 1 clinical study without the need to use radiolabel and with limited additional cost. A “human first” approach would allow for streamlining of follow up activities such as animal radiolabeled ADME studies, drug-drug interaction in vitro, and clinical studies and defining regulated bioanalysis strategy in toxicology and clinical studies to answer the key questions raised by the human data. Furthermore, 19F NMR provides an opportunity to quantitatively investigate metabolite systemic exposures at steady state, as requested by the MIST guidance. Multiple dosing of a radiolabeled tracer is not practical, which means investigation of steady-state levels typically relies on, for example, development of validated bioanalytical assays or extrapolations from single-dose data.
The need for a traditional 14C human ADME study later in development could also be questioned, potentially saving the need to expose healthy volunteers to a radioactive dose of the drug. The main limitations of the study design are sensitivity of the NMR instrument and that it is only applicable to fluorinated drugs. Whether 19F NMR would be sensitive enough to apply to a given clinical study is complex to answer. The dose of administered drug, the extent of its metabolism, the extent of drug absorption and distribution, and the rate of excretion are all factors that should be considered. Finally, the number of fluorine atoms (CF3 versus single F) will also influence the sensitivity as CF3 resonances are equivalent and give rise to a threefold more intense signal. Available PK, TK, and preclinical ADME data can be used, in addition to the proposed clinical dose, to evaluate the likelihood of success. In our experience, we estimate that, for an orally dosed drug containing a CF3 moiety, a 19F NMR study could be considered for doses above 50 mg provided factors discussed above are also taken into consideration.
Acknowledgments
The authors acknowledge Pieter Jacob Swart for contributions to discussions and for support of the study. We also thank Grazyna Ciszewska for the synthesis of [14C]BYL719 (Alpelisib) and Vincent Bordas for the synthesis of metabolite M4.
Authorship Contributions
Participated in research design: James, Marvalin, Luneau, Meissner, and Camenisch.
Conducted experiments: Marvalin and Luneau.
Performed data analysis: James, Marvalin, and Luneau.
Wrote or contributed to the writing of the manuscript: James, Marvalin, Luneau, and Meissner.
Footnotes
- Received February 13, 2017.
- Accepted May 22, 2017.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- DRM
- drug-related material
- hADME
- human absorption, distribution, metabolism and excretion study
- HPLC
- high-performance liquid chromatography
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- LSC
- liquid scintillation counting
- PK
- pharmacokinetics
- Copyright © 2017 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}