


Abstract
Primary human hepatocytes in culture are commonly used to evaluate cytochrome P450 (P450) induction via an enzyme activity endpoint. However, other processes can confound data interpretation. To this end, the impact of time-dependent P450 inhibition in this system was evaluated. Using a substrate-cassette approach, P450 activities were determined after incubation with the prototypic inhibitors tienilic acid (CYP2C9), erythromycin, troleandomycin, and fluoxetine (CYP3A4). Kinetic analysis of enzyme inactivation in hepatocytes was used to describe the effect of these time-dependent inhibitors and derive the inhibition parameters kinact and KI, which generally were in good agreement with the values derived using recombinant P450s and human liver microsomes (HLMs). Tienilic acid selectively inhibited CYP2C9-dependent diclofenac 4′-hydroxylation activity, and erythromycin, troleandomycin, and fluoxetine inhibited CYP3A4-dependent midazolam 1′-hydroxylation in a time- and concentration-dependent manner. Fluoxetine also inhibited CYP2C19-dependent S-mephenytoin 4′-hydroxylation in a time- and concentration-dependent manner in hepatocytes, HLMs, and recombinant CYP2C19 (KI 0.4 μM and kinact 0.5 min–1). As expected, the effect of fluoxetine on CYP2D6 in hepatocytes was consistent with potent yet reversible inhibition. A very weak time-dependent CYP2C9 inhibitor (AZ1, a proprietary AstraZeneca compound; KI 30 μM and kinact 0.02 min–1) effectively abolished CYP2C9 activity over 24 h at low (micromolar) concentrations in primary cultured human hepatocytes. This work demonstrates that caution is warranted in the interpretation of enzyme induction studies with metabolically stable, weak time-dependent inhibitors, which may have dramatic inhibitory effects on P450 activity in this system. Therefore, in addition to enzyme activity, mRNA and/or protein levels should be measured to fully evaluate the P450 induction potential of a drug candidate.
Inhibition of cytochrome P450 (P450)-dependent metabolism is a prevalent source of drug-drug interactions (DDIs) and may result in serious clinical consequences (Jankel and Fitterman, 1993; Bertz and Granneman, 1997) via either reversible or irreversible means. An assessment of the potential of a new chemical entity to cause DDIs via inhibition of P450 metabolism is important early in the drug discovery process. Mechanism-based P450 inhibitors can be characterized as displaying NADPH-, concentration-, and time-dependent quasi-irreversible or irreversible inactivation because of chemical modification of the heme and/or protein, and the inactivation rate is diminished in the presence of a competing substrate (Silverman, 1998). Such inhibitors may be of specific concern; first because the in vivo inhibitory effect lasts longer than that for reversible inhibitors and inactivated P450 has to be replaced by newly synthesized protein; and second because of an increased toxicological risk via the generation of reactive species (Zhou et al., 2005).
Many drugs are mechanism-based inhibitors of P450, including the macrolide antibiotics erythromycin and troleandomycin (Larrey et al., 1983), tienilic acid (Lopez-Garcia et al., 1994), and fluoxetine (Mayhew et al., 2000). Thus, in vitro screens to determine both the degree and nature of P450 inhibition (typically determining the time dependence of inhibition) are now used across the pharmaceutical industry. More often than not, these assays focus on the five major drug-metabolizing P450s in humans: 1A2, 2C9, 2C19, 2D6, and 3A4 and are studied using one or a combination of the following tools; recombinantly expressed P450s, human liver microsomes, and human hepatocytes.
Arguably, human hepatocytes provide the closest in vitro model to human liver by providing the full complement of xenobiotic-metabolizing enzymes and transporters. With the improvement of cryopreservation techniques, human hepatocytes are increasingly used throughout the industry for assessing the metabolic stability of new chemical entities (McGinnity et al., 2004) and, more recently, for the study of both reversible inhibition of CYP2C9 (McGinnity et al., 2005) and time-dependent inhibition of CYP3A4 (Ring et al., 2005; Zhao et al., 2005). Hepatocytes in primary culture are widely recognized as being the gold standard for predicting P450 induction in both preclinical species and humans (LeCluyse et al., 2000; Luo et al., 2004; Kato et al., 2005). Induction of P450s can be demonstrated using one or more of a combination of different endpoints: mRNA, protein via Western blotting, and activity using a selective substrate. Although regulation of P450 activity in vivo is the key parameter with respect to DDIs, caution must be used when interpreting data from primary cultures based solely on an in vitro activity readout, since it is recognized that mechanism-based inhibitors of P450s may lead to a decrease in activity that will be independent of any increase in activity caused by induction (Pichard et al., 1990; Silva and Nicoll-Griffith, 2002; Ring et al., 2005).
The efficiency of time-dependent inhibition is a function of the ratio of kinact (maximal inactivation rate constant) to KI (inhibitor concentration that supports half the maximal rate of inactivation), both of which can be determined from in vitro experiments. Approaches to predicting the in vivo DDI potential of time-dependent P450 inhibitors have ranged from a relatively simplistic approach using kinact and KI values, the enzyme degradation rate (kdeg) and a single inhibitor concentration (Mayhew et al., 2000; Wang et al., 2004), to a more complex, physiological-based model, incorporating the changing concentrations of inhibitor and substrate (Ito et al., 2003).
In this study, the impact of time-dependent P450 inhibition in human hepatocytes in primary culture has been evaluated, using a substrate cassette strategy. An adaptation of the physiological-based model developed by Ito et al. (2003) was used to describe the effect on P450 activities observed in primary culture by prototypic time-dependent P450 inhibitors. KI and kinact values determined in cultured human hepatocytes were compared with values obtained from rP450s and HLMs. The additional value of using primary hepatocytes for the assessment of coincidental time-dependent inhibition and P450 induction is also developed and discussed.
Materials and Methods
Materials. Diclofenac, 4′-hydroxydiclofenac, (±)fluoxetine, phenacetin, paracetamol, and β-nicotinamide adenine dinucleotide phosphate reduced form (β-NADPH), phosphate-buffered saline tablets, and Tween 20 were purchased from Sigma-Aldrich Chemical (Gillingham, UK) and were of the highest grade available. Bufuralol, S-mephenytoin, and midazolam were purchased from Sequoia Research Products Ltd. (Oxford, UK). Hydroxybufuralol, 4′-hydroxymephenytoin, and 1′-hydroxymidazolam were purchased from Ultrafine Chemicals (Manchester, UK). Dimethyl sulfoxide (DMSO) and trichloroacetic acid were purchased from Fisher Scientific (Loughborough, UK), and methanol was purchased from Romil Ltd. (Cambridge, UK). Hepatocyte maintenance media (HMM) and HMM SingleQuot supplement containing dexamethasone (final concentration 50 nM), gentamicin (50 μg/ml), and amphotericin B (50 ng/ml) were obtained from Cambrex Bioscience (Wokingham, UK); ITS+ supplement containing insulin (final concentration 6.25 μg/ml), transferrin (6.25 μg/ml), selenium (6.25 ng/ml), linoleic acid (5.35 μg/ml), and bovine serum albumin (1.25 mg/ml), and Sterile 6-well Biocoat collagen type I plates were obtained from BD Biosciences (Oxford, UK).
Bactosomes prepared from Escherichia coli cells coexpressing recombinant human NADPH-P450 reductase and individual human P450s (CYP1A2, 2C9, 2C19, 2D6, and 3A4) were purchased from Cypex (Dundee, UK). Pooled human liver microsomes were purchased from BD Gentest (Bedford, MA).
Preparation of Human Hepatocytes. Human hepatocytes were prepared from an isolated lobe of human liver (obtained from local hospitals with ethical approval) using a procedure described previously (McGinnity et al., 2004). Donor demographics for the five hepatocyte donors were as follows: two Caucasian males aged 63 and 70 years and three Caucasian females age 64 to 81 years; liver was obtained via surgical resection; medical history was not available. Hepatocyte viability for the five donors, as determined by the trypan blue exclusion method, was between 86 and 98%.
Culturing of Human Hepatocytes. Freshly isolated hepatocytes were diluted to a cell concentration of approximately 8 × 106/ml using ice-cold hepatocyte suspension buffer (2.2 g of NaHCO3, 2.34 g of Na HEPES, 1 liter powder equivalent of Dulbecco's modified Eagle's medium (Sigma-Aldrich Chemical) diluted in 1 liter of water and adjusted to pH 7.4 with 1 M HCl). Cells were centrifuged (50g, 5 min, 4°C), the supernatant was removed by aspiration, and the pellet was resuspended in a small volume of sterile supplemented HMM, counted using the trypan blue exclusion method, and then further diluted to 0.75 × 106 cells/ml. Working using sterile techniques, cells were plated at a seeding density of 1.5 × 106 per well in 2 ml (0.16 × 106/cm2) and left for 16 h at 37°C, 90% relative humidity, and 5% CO2 atmosphere. Cells were washed and assays performed using supplemented HMM.
Determination of P450 Activity and CLint of Inhibitors in Cultured Human Hepatocytes. Inhibitor and substrate stocks were prepared in DMSO at 200-fold incubation concentrations. To minimize the known P450-inhibitory effects of DMSO, the final concentration in all incubations was 1% (v/v). To cultured human hepatocytes on six-well collagen-coated plates, each 200× inhibitor stock (10 μl) was added to a well containing supplemented HMM (2 ml) to give final inhibitor concentrations of 0.1, 1, and 10 μM per well. The three remaining wells from each plate were spiked with DMSO (10 μl) as controls. A separate plate was prepared in this way for each time point and inhibitor. At several time points from 0 to 48 h, 200× cassetted substrate stock (10 μl) was added to each well to give a final phenacetin concentration of 26 μM, diclofenac 9 μM, S-mephenytoin 31 μM, bufuralol 9 μM, and midazolam 3 μM. Aliquots (100 μl) were removed 5, 10, 15, 20, and 30 min after addition of probe substrate, and samples were quenched in ice-cold methanol (100 μl). Phenacetin deethylation, diclofenac 4′-hydroxylation, S-mephenytoin 4′-hydroxylation, bufuralol 1′-hydroxylation, and midazolam 1′-hydroxylation were used as selective probe reactions for CYP1A2, 2C9, 2C19, 2D6, and 3A4 activity, respectively. The P450 isoform selectivity of these reactions has been established previously (Weaver et al., 2003). All substrates were incubated at their measured Km for the relevant P450 with the exception of diclofenac/CYP2C9, which was incubated at 4 times Km to allow analytical detection of the metabolite. The formation of 4′-hydroxydiclofenac remains selective for CYP2C9 at 9 μM diclofenac (data not shown). Aliquots from each well were assayed against a metabolite standard curve for the amount of metabolite produced at each time point. The apparent rate of metabolite formation over time was compared in the presence and absence of inhibitor. P450 activity is expressed as a percentage relative to a solvent control incubation (in the absence of inhibitor) at each corresponding time point.
The initial CLint of each inhibitor in cultured human hepatocytes was established on a separate plate using a single, low substrate concentration (1 μM final concentration; 1% v/v DMSO). Aliquots (100 μl) were removed at 0, 5, 10, 20, 30, 60, and 90 min and quenched in ice-cold methanol (100 μl). In addition, to determine the concentration of inhibitor present in the incubation during the assessment of P450 activity (up to 48 h), an aliquot (100 μl) was taken from each well and quenched in ice-cold methanol (100 μl), before addition of cassetted substrate.
Samples were subsequently chilled (–20°C, 1 h) and then centrifuged (3500 rpm, 15 min, 4°C). The supernatants were removed and transferred into HPLC vials and analyzed by HPLC-MS/MS as described below. Cells were collected from the several time point samples and stored (–30°C) in phosphate buffer (0.1 M, pH 7.4) for Western blotting.
Estimation of fuinc in rCYP2C9, Human Liver Microsomes, and Human Hepatocytes. The extent of binding of compounds to rCYP2C9, human liver microsomes, and human hepatocytes was estimated using algorithms based on lipophilicity proposed by Austin et al. (2002, 2005).
Determination ofKI andkinact in rP450s and Human Liver Microsomes.KI and kinact values were determined in rP450s and human liver microsomes by an adapted version of the automated method described by Atkinson et al. (2005). Briefly, the preincubation used rP450 (25 pmol/ml) or HLMs (1 mg/ml) in phosphate-buffered saline (0.1 M, pH 7.4), NADPH (1 mM), and six different concentrations of test inhibitor in DMSO (1% v/v), which were incubated at 37°C for 3, 6, and 9 min. After a 10-fold dilution into NADPH (1 mM) and the relevant P450 substrate, the second reaction was allowed to proceed (15 min). For CYP1A2, phenacetin was used at 75 μM, for 2C9, diclofenac (10 μM), for 2C19, S-mephenytoin (100 μM), for 2D6, bufuralol (40 μM), and for 3A4, midazolam (10 μM). Aliquots (50 μl) were quenched in ice-cold methanol (100 μl) and prepared for HPLC-MS/MS as described below. Manual incubations were performed for fluoxetine using the method of Atkinson et al. (2005). Briefly, six concentrations (0.04–2 μM) of fluoxetine in DMSO (1% v/v) were incubated with rP450 (25 pmol/ml) or HLMs (1 mg/ml) in phosphate-buffered saline (0.1 M, pH 7.4), NADPH (1 mM) at 37°C for 0, 0.5, 1, 2.5, 5, and 10 min. After a 10-fold dilution into NADPH (1 mM) and S-mephenytoin (100 μM), the reaction was allowed to proceed (15 min). Aliquots (50 μl) were quenched in ice-cold methanol (100 μl) and prepared for HPLC-MS/MS as described below.
Analytical Methods. Aliquots (20 μl) were analyzed by HPLC-MS/MS for metabolite appearance of the probes of CYP1A2, 2C9, 2C19, 2D6, and 3A4 activity and parent loss of the inhibitors. Mass spectrometry was conducted on a Micromass Quattro Ultima triple quadrupole by using an Alliance HT Waters 2790 (Waters, Milford, MA) HPLC system for separation. Mass spectroscopy conditions for the five metabolites used as probes of P450 activity were as described previously (Weaver et al., 2003). A Synergi (4-μm) Max-RP C12 column (50 × 4.6 mm; Phenomenex, Cheshire, UK) and mobile phases of 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B) were used for the chromatography. The gradient was as follows: 97% A (0–0.3 min), 5% A (0.55–1.55 min), 97% A (1.6 min). The stop time was 2.5 min, the flow rate was 1.5 ml · min–1, and column temperature was 40°C.
Data Analysis. CLint estimates were determined using the rate of parent disappearance (at a single, low substrate concentration) as described previously (McGinnity et al., 2004).
KI and kinact values in rP450 and HLMs were estimated from the natural log of the percentage of control activity remaining, following incubation with a single inhibitor concentration, plotted against the time of preincubation. The slope, k, is the inactivation rate constant (describing the rate of inactivation at that inhibitor concentration). This was determined for data from all inhibitor concentrations. Nonlinear regression analysis (WinNonlin; Pharsight Corporation, Mountain View, CA) was used to determine KI and kinact from the function:  where I is the preincubation inhibitor concentration, k is the inactivation rate constant for a given I, kinact is the maximal inactivation rate constant, and KI is the inhibitor concentration, I, when the inactivation rate constant is half kinact.
where I is the preincubation inhibitor concentration, k is the inactivation rate constant for a given I, kinact is the maximal inactivation rate constant, and KI is the inhibitor concentration, I, when the inactivation rate constant is half kinact.
KI and kinact values were estimated from nonlinear regression analysis of the cultured human hepatocyte data (inhibitor concentration and active P450 concentration, Eact, at each sampling time point and intrinsic clearance, CLint, of inhibitor). For the mathematical modeling, E0 (the initial uninhibited P450 concentration) was given the nominal value of 100 (100% active enzyme), and values for Eact were likewise expressed as percentages of initial P450 activity. The differential equations were derived from simple relationships describing the clearance of inhibitor and Eact from the system (human hepatocytes). In pharmacokinetics, the clearance of a given parameter is equal to the amount given to the system divided by the total exposure of the system to the parameter. Differentiation of this function reveals that the rate of change of a small amount of the parameter is equal to its clearance multiplied by its concentration at that instant. It follows, therefore, that the rate of change of inhibitor concentration (the rate of change of amount of inhibitor divided by the incubation volume, Vinc) in the human hepatocytes is equal to the product of unbound inhibitor CLint and unbound inhibitor concentration, Iu:  where fuinc denotes the fraction of inhibitor unbound in the incubation [estimated using the algorithm based on the logP or logD7.4 of the inhibitor (Austin et al., 2005)]. The fuinc values used were 0.1, 1, 0.52, 0.62, and 1 for fluoxetine, tienilic acid, erythromycin, troleandomycin, and AZ1 (a proprietary AstraZeneca compound), respectively. The unbound inhibitor concentrations (Iu) at each time point were calculated from the measured total inhibitor concentration and fuinc. CLint values for each of the test inhibitors were calculated from the respective metabolic half-lives in the plated hepatocyte incubations. When the log observed inhibitor concentrations were not linear with respect to time, CLint values were determined only from the linear portion of the log inhibitor concentration-time profile. Use of these observed CLint values in the model would have resulted in an underestimation of inhibitor concentration at the later time points, since CLint abated with time. To ensure equivalent simulated and observed inhibitor concentrations and, therefore, ensure accurate Iu values to input into eq. 4, an exponential term, expK·t, was used. Describing eq. 2 in this way was necessary because actual inhibitor concentrations were measured only at specified time points, whereas the model requires details of the constantly changing inhibitor concentration so that the continuous alteration of active P450 concentration is conveyed. The constant, K, was determined as follows:
where fuinc denotes the fraction of inhibitor unbound in the incubation [estimated using the algorithm based on the logP or logD7.4 of the inhibitor (Austin et al., 2005)]. The fuinc values used were 0.1, 1, 0.52, 0.62, and 1 for fluoxetine, tienilic acid, erythromycin, troleandomycin, and AZ1 (a proprietary AstraZeneca compound), respectively. The unbound inhibitor concentrations (Iu) at each time point were calculated from the measured total inhibitor concentration and fuinc. CLint values for each of the test inhibitors were calculated from the respective metabolic half-lives in the plated hepatocyte incubations. When the log observed inhibitor concentrations were not linear with respect to time, CLint values were determined only from the linear portion of the log inhibitor concentration-time profile. Use of these observed CLint values in the model would have resulted in an underestimation of inhibitor concentration at the later time points, since CLint abated with time. To ensure equivalent simulated and observed inhibitor concentrations and, therefore, ensure accurate Iu values to input into eq. 4, an exponential term, expK·t, was used. Describing eq. 2 in this way was necessary because actual inhibitor concentrations were measured only at specified time points, whereas the model requires details of the constantly changing inhibitor concentration so that the continuous alteration of active P450 concentration is conveyed. The constant, K, was determined as follows:  where CLint, initial was the CLint established from the linear portion of the log [inhibitor]-time profile and t was every time point at which the inhibitor concentration was determined. The values for K used in the nonlinear regression to estimate KI and kinact were –0.006 for fluoxetine and tienilic acid, –0.002 for erythromycin, and 0 for troleandomycin and AZ1. The merit of using these values in the model was assessed by goodness of fit measurements for Eact (Akaike criteria and value and spread of residuals).
where CLint, initial was the CLint established from the linear portion of the log [inhibitor]-time profile and t was every time point at which the inhibitor concentration was determined. The values for K used in the nonlinear regression to estimate KI and kinact were –0.006 for fluoxetine and tienilic acid, –0.002 for erythromycin, and 0 for troleandomycin and AZ1. The merit of using these values in the model was assessed by goodness of fit measurements for Eact (Akaike criteria and value and spread of residuals).
The rate of change of Eact is similarly expressed (eq. 4; but takes account of the resynthesis of active P450). The clearance of Eact in the kinetic model equals the sum of Eact entering (synthesis) and leaving (inactivation) the compartment. As with eq. 2, a rate of change of concentration rather than amount is described, but no incubation volume term appears because the clearance is written as a sum of rate constants (the rate constant for time-dependent inactivation being kinact × I/(KI + I), where clearance equals the product of rate constant and volume. It is also important to note that E0 – Eact, rather than Eact, is the “missing” concentration of active P450 driving the resynthesis:  where kinact, KI, E0, kdeg, and fuinc represent the maximal inactivation rate constant, the inhibitor concentration when the inactivation rate constant is kinact/2, the initial concentration of P450, and the degradation or turnover rate constant of the inhibited P450. In the absence of definitive degradation rate constants for individual P450s in cultured human hepatocytes, unless stated, a value of 0.00025 min–1 was used for all P450s, based on the turnover rate constant for CYP3A in cultured human hepatocytes (Pichard et al., 1992). In the mathematical model described here, it was assumed that the degradation and synthesis rate constants were equal and that this was unaffected by the presence of P450 inhibitors.
where kinact, KI, E0, kdeg, and fuinc represent the maximal inactivation rate constant, the inhibitor concentration when the inactivation rate constant is kinact/2, the initial concentration of P450, and the degradation or turnover rate constant of the inhibited P450. In the absence of definitive degradation rate constants for individual P450s in cultured human hepatocytes, unless stated, a value of 0.00025 min–1 was used for all P450s, based on the turnover rate constant for CYP3A in cultured human hepatocytes (Pichard et al., 1992). In the mathematical model described here, it was assumed that the degradation and synthesis rate constants were equal and that this was unaffected by the presence of P450 inhibitors.
Eact values were determined using the P450-selective probe substrates (CYP2C9-dependent diclofenac 4′-hydroxylation, CYP2C19 S-mephenytoin 4′-hydroxylation, CYP2D6 bufuralol 1′-hydroxylation, and CYP3A4 midazolam 1′-hydroxylation) but were modeled as percentages of Eo. Equations 2 and 4 were replicated for each additional concentration of inhibitor used (for example, four differential equations were used to model the data and estimate KI and kinact when two inhibitor concentrations were used).
In Vivo Simulation of P450 Activity after Dosing with AZ1. Using the methodology of Ito et al. (2003) and based on expected human pharmacokinetics and therapeutic dose, CYP2C9 activity in human liver was simulated after oral dosing with AZ1 (70 mg every 24 h for 72 h). Dose was 200 μmol, Vmax 65 μmol · min–1, km 50 μM, KP1 (liver to blood concentration of AZ1) 1, fb (unbound fraction of AZ1 in blood) 0.01, ka (first-order absorption rate constant) 0.01 min–1, Fa (fraction absorbed from gastrointestinal tract) 1, Fg (intestinal availability) 1, Vsys volume of distribution in the central compartment 14,000 ml, CLr (renal clearance) 0, CYP2C9 KI 30 μM, and kinact 0.02 min–1.
Results
Determination of Inhibitor CLint and P450 Activity in Cultured Human Hepatocytes. Estimates of CLint for the P450 inhibitors, tienilic acid, erythromycin, troleandomycin, fluoxetine, and a proprietary AstraZeneca compound (AZ1) were determined in cultured human hepatocytes from five individual donors (Table 1). Assays were carried out 12 to 16 h after plating and CLint values were determined by the rate of parent disappearance over 90 min.
CLint values of inhibitors determined in five human hepatocyte donors
Assays were carried out 12 to 16 h after plating and CLint values were determined by the rate of parent disappearance over 90 min as described under Materials and Methods. Final concentration of compounds was 1 μM. Due to limited numbers of hepatocytes, all compounds were not incubated in all donors.
The activity of the five major human hepatic P450s (CYP1A2, 2C9, 2C19, 2D6, and 3A4) was assessed in cultured human hepatocytes by use of a cocktail of P450-specific substrates, which allowed for the maximum amount of data to be retrieved from the limited resource of human hepatocytes.
Time-Dependent Inhibition of P450 Activity in Cultured Human Hepatocytes. The limited number of hepatocytes from each individual donor constrained the design of the time course assays. Each inhibitor was incubated with hepatocytes from a minimum of three individual donors over a 48-h time course. The absolute activity in the absence of inhibitor, for the major isoforms studied, CYP2C9 and CYP3A4, changed less than 2-fold during the course of the 48-h incubation. To control for these changes in activity over the time course of the experiment, P450 activity was expressed as a percentage relative to a solvent control at each time point. Using mean CLint and percentage P450 activity values determined from the human hepatocytes and a P450 turnover constant (kdeg) of 0.00025 min–1, as determined for CYP3A4 in cultured human hepatocytes (Pichard et al., 1992) the mean KI and kinact values were estimated from nonlinear regression analysis using eqs. 2 and 4.
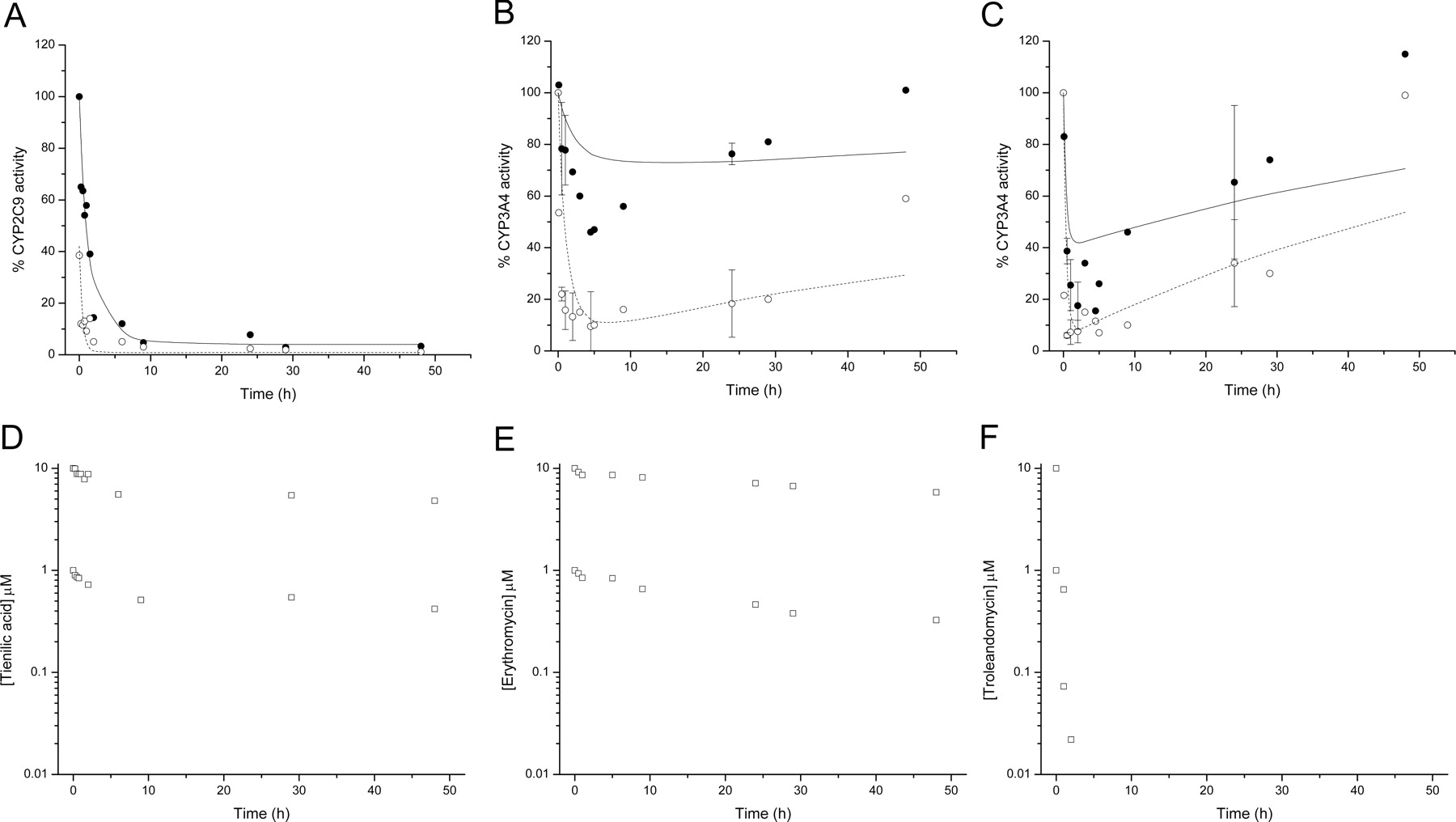
P450 activity in cultured human hepatocytes after incubation with tienilic acid, erythromycin, and troleandomycin. Using cultured human hepatocytes, CYP2C9-dependent diclofenac 4′-hydroxylation after incubation with tienilic acid (A), and CYP3A4-dependent midazolam 1′-hydroxylation after incubation with erythromycin (B) and troleandomycin (C) were determined as described under Materials and Methods. Data points represent the mean activity relative to a solvent control incubation (in the absence of inhibitor) at the corresponding time point from donors 1, 3, and 5 for tienilic acid, and donors 1, 2, 4, and 5 for erythromycin and troleandomycin. Where activities from at least three donors were measured, error bars reflect the standard deviation from the mean. Closed and open circles indicate incubation with 1 μM and 10 μM inhibitor, respectively. The solid and dashed lines indicate nonlinear regression of the 1 and 10 μM inhibitor data, respectively, using eq. 4 as described under Materials and Methods. Open squares represent the mean concentration of tienilic acid (D), erythromycin (E), and troleandomycin (F), from the 1 and 10 μM incubations, over the time course of the experiment as determined by HPLC-MS/MS.
Fitted inactivation curves for the different concentrations of inhibitors against the individual P450 are compared with the experimental data in Figs. 1, 2, 3, and 4.
Figure 1 (A–C) shows mean P450 activities in cultured human hepatocytes over time after incubation with prototypic mechanism-based inhibitors tienilic acid, erythromycin, and troleandomycin, respectively. Tienilic acid inhibited CYP2C9-mediated diclofenac 4′-hydroxylation in a time- and concentration-dependent manner (Fig. 1A). During incubation with 1 μM tienilic acid, maximal inhibition (to 5% of control activity) was reached at 9 h postincubation, and this degree of inhibition was maintained for at least 48 h. Tienilic acid at 10 μM effectively abolished CYP2C9 activity for the duration of the experiment. The activities of CYP1A2, 2C19, 2D6, and 3A4 were unaffected by the presence of up to 10 μM tienilic acid. Erythromycin inhibited CYP3A4-mediated midazolam 1′-hydroxylation in a time- and concentration-dependent manner (Fig. 1B). After 5 h of incubation, erythromycin at 1 and 10 μM caused maximal CYP3A4 inhibition (to 46 and 10% of control activity, respectively) and by 48 h, 100 and 59% activity, respectively, was restored. The activities of CYP1A2, 2C9, 2C19, and 2D6 were unaffected by the presence of up to 10 μM erythromycin. Maximal CYP3A4 inhibition (to 17% of control activity) was reached approximately 5 h into an incubation with 1 μM troleandomycin (Fig. 1C), whereas at 10 μM, CYP3A4 activity was effectively abolished by 0.5 h, an inhibition level maintained up to 9 h. Full CYP3A4 activity for both concentrations was restored by 48 h. The activities of CYP1A2, 2C9, 2C19, and 2D6 were unaffected by the presence of up to 10 μM troleandomycin. Mean inhibitor concentrations throughout the time course of the assay were determined (Fig. 1, D–F). Troleandomycin was essentially cleared after approximately 3 h (Fig. 1F), whereas measurable levels of tienilic acid and erythromycin were maintained for the duration of the incubation (Fig. 1, D and E).

Simulation of P450 activity in cultured human hepatocytes after incubation with troleandomycin using three different CYP3A4 turnover rate constants. Data points represent the mean CYP3A4-dependent midazolam 1′-hydroxylation activity, relative to a solvent control incubation at the corresponding time point, after incubation with 1 μM (closed circles) and 10 μM (open circles) troleandomycin as shown in Fig. 1C. The solid and dashed lines indicate nonlinear regression of the 1 and 10 μM inhibitor data, respectively, using eq. 4 with kdeg of 0.00025 min–1 (A), 0.0005 min–1 (B), and 0.00075 min–1 (C).
Figure 2 shows the effect of using different kdeg values in eq. 4 by plotting CYP3A4 activity against time of incubation with troleandomycin with fitted inactivation curves based on using kdeg values of 0.00025 min–1 (t1/2 = 46 h) (Fig. 2A), 0.0005 min–1 (t1/2 = 23 h) (Fig. 2B), and 0.00075 min–1 (t1/2 = 15 h) (Fig. 2C). Clearly, the rate of regain of activity is a function of kdeg, yet over the range used, kdeg has little effect on the estimates of KI and kinact (data not shown).
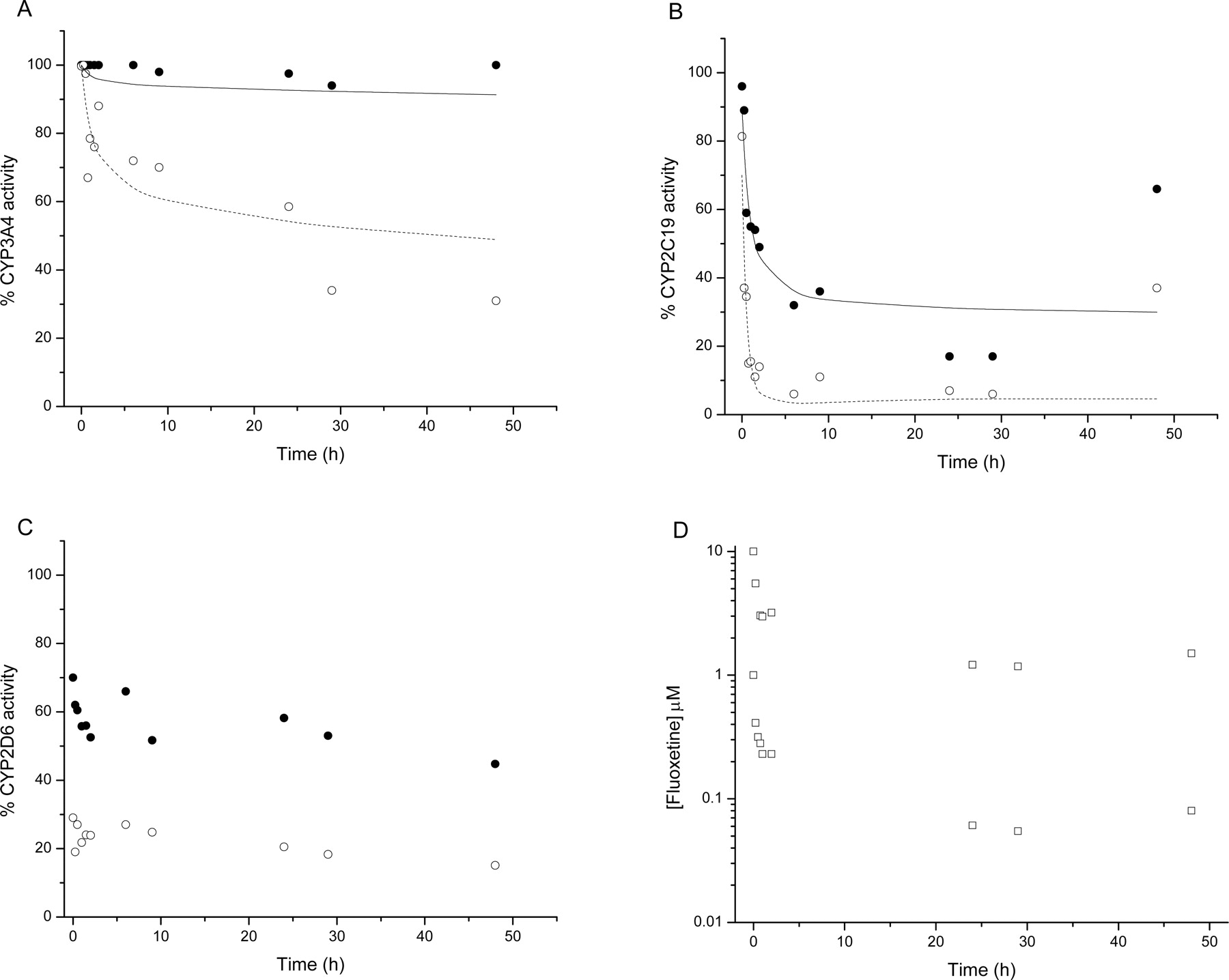
Figure 3, A to C, shows plots of CYP3A4, 2C19, and 2D6 activity, respectively, against time of incubation with fluoxetine. Fluoxetine clearly inhibited CYP3A4-mediated midazolam 1′-hydroxylation and CYP2C19-mediated S-mephenytoin 4′-hydroxylation in a time- and concentration-dependent manner. Figure 3A shows that CYP3A4 activity was not significantly inhibited by 1 μM fluoxetine yet reached maximal inhibition 48 h after incubation with 10 μM fluoxetine (to 31% of control activity). Figure 3B shows that CYP2C19 activity was maximally inhibited (to 18% of control activity) between 24 and 29 h during incubation with 1 μM fluoxetine, and 66% of activity was restored by 48 h. Fluoxetine at 10 μM effectively abolished CYP2C19 activity by 6 h with 38% of activity being restored by 48 h. CYP2D6-mediated bufuralol 1′-hydroxylation was inhibited to approximately 60 and 25% of control activity by 1 and 10 μM fluoxetine, respectively (Fig. 3C). After 48 h of incubation, CYP2D6 activity was reduced to 48 and 16% of control activity by 1 and 10 μM fluoxetine, respectively. CYP1A2 and 2C9 activities were unaffected by the presence of up to 10 μM fluoxetine. Fluoxetine concentration was determined throughout the time course of the assay (Fig. 3D).
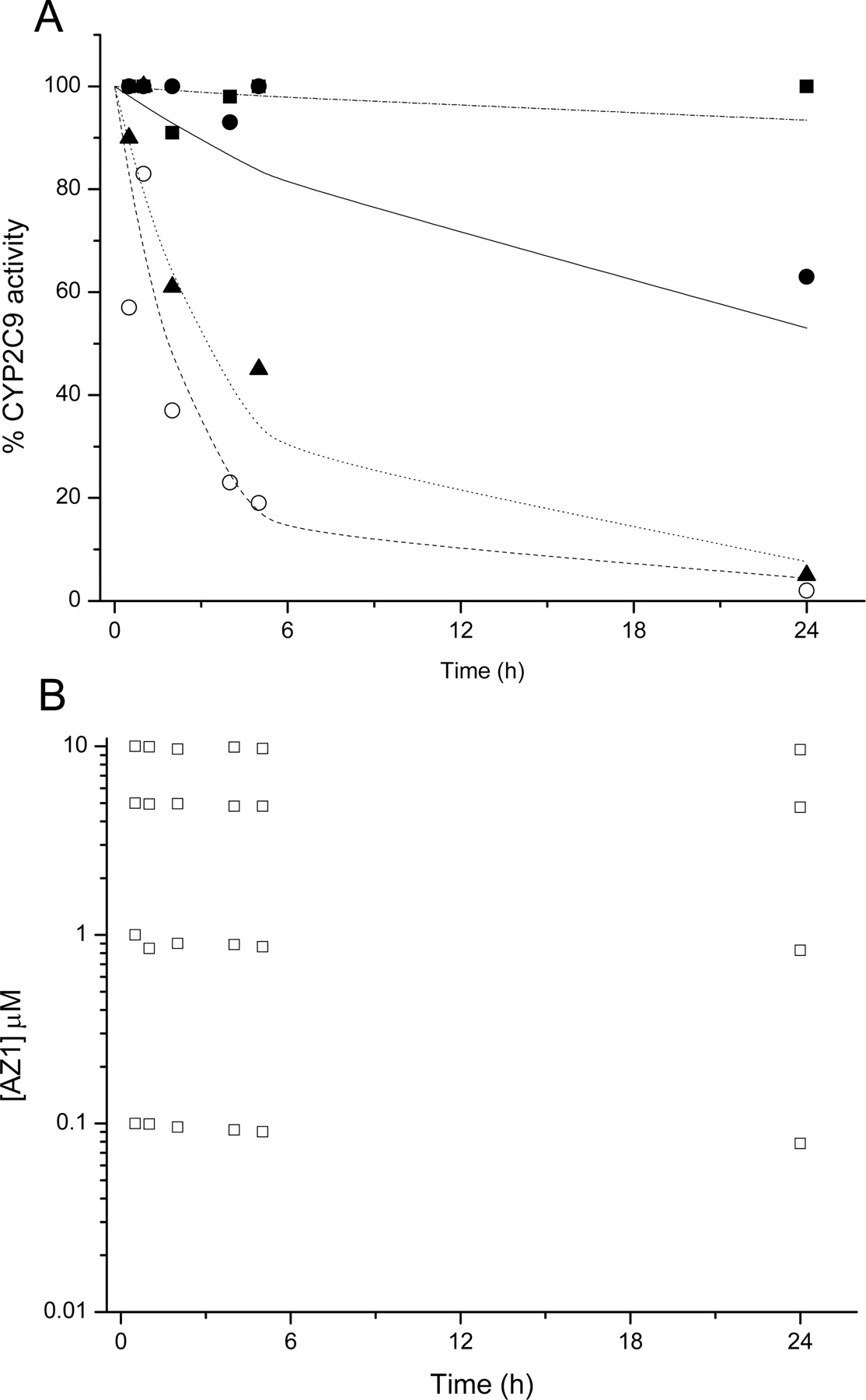
Figure 4A plots CYP2C9 activity against time of incubation with a proprietary AstraZeneca compound, AZ1. AZ1 inhibited CYP2C9-mediated diclofenac 4′-hydroxylation in a time- and concentration-dependent manner from 0.1 to 10 μM. CYP2C9 activity was not significantly inhibited by 0.1 μM AZ1 but was effectively abolished by 24 h after incubation with 5 μM AZ1. CYP1A2, 2C19, 2D6, and 3A4 activities were unaffected by the presence of up to 10 μM AZ1. AZ1 concentration was determined throughout the time course of the assay (Fig. 4B).
Comparison ofKI andkinact Values from Different Enzyme Sources. The kinetic constants of time-dependent inhibition, KI and kinact, were estimated in rP450s for the inhibitor/P450 pair that demonstrated clear time-dependent inhibition in human hepatocytes. KI and kinact values were determined using rP450s and HLMs as described under Materials and Methods. Table 2 compares KI and kinact estimates generated in rP450s and human hepatocytes with the relevant selective substrate to data reported in the literature, using HLMs or rP450s as the enzyme source. Generally, there is good agreement between values generated in this laboratory and those reported in the literature. Mean values from triplicate rP450 experiments were compared with KI and kinact estimates generated using mean human hepatocyte data shown in Figs. 1, 3, and 4. The fraction unbound (fuinc) of the inhibitors in the hepatocyte milieu were estimated from the method of Austin et al. (2005) and incorporated in the model to generate unbound KI values (see Materials and Methods), allowing a direct comparison to values estimated from rP450 where, due to the very low protein level (<0.05 mg/ml), values approaching unbound KI would be determined. Table 2 lists both apparent and unbound KI values for fluoxetine in HLMs, where fuinc at 1 mg/ml HLM is 0.1 (Austin et al., 2002), which is of the same order as that determined experimentally (Margolis and Obach, 2003). All other inhibitors, being significantly less lipophilic than fluoxetine, have higher fuinc and, therefore, apparent KI approaches unbound KI values.
Kinetic parameters of time-dependent inhibitors in different in vitro matrices
Tienilic acid at CYP2C9 generated KI and kinact values from rP450 versus human hepatocytes of 2 μM and 0.19 min–1 versus 2 μM and 0.05 min–1, respectively; erythromycin/CYP3A4 of 9 μM and 0.12 min–1 versus 11 μM and 0.07 min–1; troleandomycin/CYP3A4 of 0.3 μM and 0.12 min–1 versus 0.4 μM and 0.05 min–1; and AZ1/CYP2C9 of 30 μM and 0.02 min–1 versus 19 μM and 0.02 min–1. Therefore, for tienilic acid, erythromycin, troleandomycin, and AZ1, there was good concordance of the kinetic constants between the rP450s and human hepatocytes.
For fluoxetine, time-dependent inhibition in human hepatocytes was observed for both CYP3A4- and CYP2C19-mediated reactions. There was good agreement between unbound KI and kinact values generated using rCYP3A4 (2 μM and 0.03 min–1, respectively), HLM [0.5 μM (apparent KI 5 μM) and 0.01 min–1], and human hepatocytes (1 μM and 0.01 min–1) for midazolam 1′-hydroxylation. The apparent KI and kinact values generated in HLM similarly agreed with those previously reported, 5 μM and 0.02 min–1, respectively (Mayhew et al., 2000). The time-dependent inhibition of rCYP2C19 by fluoxetine, using S-mephenytoin 4′-hydroxylation as the probe reaction, is shown in Fig. 5. There was a reasonable agreement for the unbound KI and kinact values generated using HLMs [0.8 μM (apparent KI 8 μM) and 0.03 min–1] and human hepatocytes (0.2 μM and 0.04 min–1). Using rCYP2C19, the unbound KI (0.4 μM) was similar to that of the HLM and hepatocyte values, but the kinact was significantly higher (0.5 min–1) (Table 2). No time- or NADPH-dependent inhibition was observed with fluoxetine for rCYP2D6 in accordance with the lack of significant time dependence observed for flouxetine/CYP2D6 in human hepatocytes (Fig. 3C).

CYP3A4, 2C19, and 2D6 activity in cultured human hepatocytes after incubation with fluoxetine. Using cultured human hepatocytes, CYP3A4-dependent midazolam 1′-hydroxylation (A), CYP2C19-dependent S-mephenytoin 4′-hydroxylation (B), and CYP2D6-dependent bufuralol 1′-hydroxylation (C) were determined after incubation with 1 μM (closed circles) and 10 μM (open circles) fluoxetine, as described under Materials and Methods. Data points represent the mean activity relative to a solvent control incubation at the corresponding time point, from donors 1, 3, and 5. Where activities from at least three donors were measured, error bars reflect the standard deviation from the mean. The solid and dashed lines indicate nonlinear regression of the 1 and 10 μM fluoxetine data, respectively, using eq. 4, as described under Materials and Methods. Open squares represent the mean concentrations of fluoxetine (D) from the 1 and 10 μM incubations over the time course of the experiment as determined by HPLC-MS/MS.
Discussion
Drug discovery departments in the pharmaceutical industry use a battery of screens to assess the potential of new chemical entities to cause DDIs via P450 inhibition and induction. Recently, screens that identify mechanism-based or, more specifically, time-dependent P450 inhibitors have been developed, in our laboratory (Atkinson et al., 2005) and others (Lim et al., 2005; Zhao et al., 2005). Such assays typically use HLMs or rP450s as the enzyme source. More recently, cryopreserved human hepatocytes in suspension have been used to study the time-dependent inactivation of CYP3A (Zhao et al., 2005).
In this study, the impact of time-dependent P450 inhibition in primary cultures of human hepatocytes was evaluated using a cocktail of P450 substrates. Use of a substrate cocktail allows for the maximum amount of data to be retrieved from the limited resource of human hepatocytes (Mohutsky et al., 2005). P450 mRNA expression and catalytic activities are differentially expressed during human hepatocyte culture (LeCluyse, 2001). In this study the effect of compounds on P450 activity are relative to a solvent control at each time point, and so the observed inhibitory effect can be assumed to be exclusively as a result of the inhibitor and not compromised by any loss of P450 enzyme throughout the culture period.
An adaptation of the physiological-based model developed by Ito et al. (2003) for predicting clinical mechanism-based DDIs was used to describe the effect on P450 activities observed in primary culture by prototypic mechanism-based P450 inhibitors. KI and kinact values were estimated from nonlinear regression analysis using eqs. 2 and 4 with an estimate of inhibitor CLint and P450 activity values determined from cultured human hepatocytes, and were compared with values obtained from rP450s and HLMs. In the absence of definitive Kdeg values for individual P450s in cultured human hepatocytes, a value of 0.00025 min–1 was used for all P450s, based on the turnover rate constant for CYP3A in cultured human hepatocytes (Pichard et al., 1992).

CYP2C9 activity in cultured human hepatocytes after incubation with AZ1. CYP2C9-dependent diclofenac 4′-hydroxylation after incubation with AZ1 was determined after incubation with 0.1 μM (closed squares), 1 μM (closed circles), 5 μM (closed triangles), and 10 μM (open circles) AZ1, as described under Materials and Methods. Data points represent the mean activity relative to a solvent control incubation at the corresponding time point, from donors 3 and 5. The dash-dot, solid, dotted, and dashed lines indicate nonlinear regression of the 0.1, 1, 5, and 10 μM AZ1 data, respectively, using eq. 4, as described under Materials and Methods. Open squares represent the mean concentrations of AZ1 (B) from the 0.1, 1, 5, and 10 μM incubations over the time course of the experiment as determined by HPLC-MS/MS.
Tienilic acid is activated by human liver CYP2C9, to form reactive thiophene sulfoxides leading to CYP2C9 inactivation (Lopez-Garcia et al., 1994). The mean KI and kinact values estimated from nonlinear regression analysis of the human hepatocyte data using a selective CYP2C9 probe (KI 2 μM and kinact 0.05 min–1) are in reasonable concordance with the values derived using rCYP2C9 (KI 2 μM and kinact 0.19 min–1) (Table 2). In addition, the fitted inactivation curves well describe the tienilic acid-dependent CYP2C9 inhibition (Fig. 1A).
The macrolide antibiotics erythromycin and troleandomycin are well established mechanism-based inhibitors of CYP3A4 via reactive nitroso species-derived heme complexes (Periti et al., 1992). Recently, the time-dependent nature of CYP3A4 inhibition by erythromycin and troleandomycin in human hepatocytes has been reported (Zhao et al., 2005), and this has been reproduced in this study. The KI and kinact values estimated from human hepatocyte data using a selective CYP3A4 probe (erythromycin, KI 11 μM and kinact 0.07 min–1; troleandomycin, 0.4 μM and 0.05 min–1, respectively) are similar to those determined in rCYP3A4 (erythromycin, 9 μM and 0.12 min–1; troleandomycin, 0.3 μM and 0.12 min–1, respectively). The values generated are also comparable to several literature reports of KI and kinact estimates using rCYP3A4 and HLMs (Table 2).
The fitted CYP3A4 inactivation curves for erythromycin and troleandomycin in human hepatocytes describe the initial degree of CYP3A4 inactivation reasonably well (Fig. 1, B and C). Although troleandomycin is a relatively potent time-dependent CYP3A4 inhibitor, it is rapidly cleared in cultured human hepatocytes (CLint values range from 37 to 51 μl/min/106 cells; Table 1, Fig. 1F), and the model simulates the recovery of CYP3A4 activity after 2 to 3 h, when essentially troleandomycin is removed from the incubation (Fig. 1F). The rate of regain of activity, described by the model, is a function of the turnover or Kdeg constant. This compares to erythromycin which, although a weaker CYP3A4 time-dependent inhibitor than troleandomycin (Table 2), is cleared more slowly (5–15 μl/min/106 cells; Fig. 1E), and the CYP3A4 activity in human hepatocytes is, therefore, simulated to recover only after several hours into the incubation (Fig. 1B). Tienilic acid is an example of a slowly cleared (5–9 μl/min/106 cells; Fig. 1D), relatively potent, time-dependent inhibitor of CYP2C9 (Table 2), which demonstrates no recovery of CYP2C9 activity over the 48 h of the incubation and is correctly simulated by the model (Fig. 1A).
The model assumes the P450 degradation rate is equal to the synthesis rate in cell culture for the duration of the experiment and describes activity recovery as a function of clearance of the irreversible inhibitor and synthesis of new protein. Of course, the situation for some inhibitors is likely more complex than that described by the model; for example, both parent and any subsequently formed metabolites may be reversibly or irreversibly inhibitory to different extents toward the P450. For both erythromycin and troleandomycin, the model underestimates the degree of regain in activity up to 48 h (Fig. 1, B and C). This may also be explainable if the half-life of CYP3A4 in these cultured human hepatocytes is somewhat shorter than previously estimated (kdeg = 0.00025 min–1, t1/2 = 46 h) (Pichard et al., 1992) as demonstrated by the simulations for troleandomycin (Fig. 2). Similarly, the underestimation could be a result of up-regulation of CYP3A4 by the macrolide antibiotics. There are apparently conflicting literature reports regarding the effects of the macrolide antibiotics on CYP3A4 mRNA, protein, and activity. Troleandomycin (20 μM) is a potent activator of human pregnane X receptor in a reporter gene assay but did not induce CYP3A4 mRNA, protein, or activity in human hepatocytes (Luo et al., 2002; Faucette et al., 2004). However, several laboratories have reported moderately raised CYP3A4 protein levels using troleandomycin (Meunier et al., 2000; Johnson et al., 2005), associated with increased (Ledirac et al., 2000) or decreased CYP3A activity (Pichard et al., 1990). Similarly, for erythromycin, there are reports of the absence (Ledirac et al., 2000) and presence (Pichard et al., 1990) of CYP3A4 protein induction as determined by Western blotting. In this laboratory, we have demonstrated erythromycin and troleandomycin to be weak inducers of CYP3A4 as determined by reverse transcription-polymerase chain reaction and Western blotting (data not shown). Despite the lack of consensus, the macrolide antibiotics are likely to be weak CYP3A4 inducers, and by using enzyme activity as the sole endpoint, time-dependent inhibition obfuscates a thorough assessment of their P450-mediated DDI potential.

Time-dependent inhibition of rCYP2C19 by fluoxetine. The data were generated using S-mephenytoin 4′-hydroxylation in rCYP2C19 as described under Materials and Methods. A, the natural log of the percentage of control activity remaining following incubation with increasing fluoxetine concentrations was plotted against the preincubation time. B, the slopes, k (inactivation rate constant), together with the fluoxetine concentration were used to determine KI and kinact by nonlinear regression analysis, using k = kinact · I /(I + KI). Each data point represents the mean from four separate experiments and error bars reflect the mean standard error.
Fluoxetine is moderately cleared in human hepatocytes, with CLint values ranging from 9 to 35 μl/min/106 cells (Table 1). Fluoxetine has been established as a mechanism-based inhibitor of CYP3A4 (Mayhew et al., 2000) and a potent yet reversible inhibitor of CYP2D6 (Bertelsen et al., 2003). The results in this study are consistent with these reports. Due to the high lipophilicity of fluoxetine (logP 4.1), it shows a significant degree of nonspecific binding to HLMs (Margolis and Obach, 2003) and hepatocytes under the experimental conditions used (Austin et al., 2002, 2005), much more so than the other inhibitors, where apparent KI will approximate unbound KI. The fuinc value is an input to the hepatocyte model, allowing unbound KI values to be estimated that are comparable to the unbound KI values estimated in rP450, in which, due to the low protein concentration, fuinc will approach unity. There was clear time dependence of fluoxetine-dependent CYP3A4 inhibition in human hepatocytes with inactivation curves for the 1 and 10 μM data described well using eq. 4 (Fig. 3A). The CYP3A4-dependent KI and kinact values in human hepatocytes (6 μM and 0.01 min–1, respectively) are comparable to values determined in rCYP3A4 (2 μM and 0.03 min–1) and HLMs (5 μM and 0.01 min–1) (Table 2). Fluoxetine at 1 and 10 μM showed significant inhibition of CYP2D6-mediated bufuralol 1′-hydroxylase activity but demonstrated only weak, if any, time dependence, consistent with reversible CYP2D6 inhibition (Fig. 3C).
Fluoxetine has been reported as an inhibitor of CYP2C19-mediated S-mephenytoin 4′-hydroxylase activity in HLMs with a Ki of 5 μM (Kobayashi et al., 1995). The previous authors used Lineweaver-Burk analysis of S-mephenytoin 4′-hydroxylase inhibition at a single time point to conclude that fluoxetine was acting competitively. However, any time dependence to the inhibition would not be apparent from such an analysis. Data generated here, in human hepatocytes, strongly suggest that this interaction is time-dependent (Fig. 3B). This was subsequently confirmed by experiments carried out in rCYP2C19 and HLMs (Table 2, Fig. 4). After consideration of nonspecific binding, the unbound KI in human hepatocytes (0.2 μM) was comparable to those determined in rCYP2C19 (0.4 μM) and HLMs (0.8 μM; Table 2). Interestingly, the kinact appears to be significantly higher when determined in rCYP2C19 (0.5 min–1) compared with HLMs and human hepatocytes (0.03 and 0.04 min–1, respectively). A similar phenomenon has been described with propranolol and CYP2D6, causing mechanism-based inhibition in rP450 but not CYP2D6 in HLMs (Palamanda et al., 2005). Although the model accurately simulated the initial loss of CYP2C19-dependent activity in human hepatocytes at the two fluoxetine concentrations, the recovery of activity at 48 h (66% at 1 μM and 38% at 10 μM) was not predicted. Reasons for this P450-specific increase in activity (it was not observed for any of the remaining four P450s studied) may include the kdeg of CYP2C19 being significantly greater than the CYP3A value of 0.00025 min–1, or the possibility that fluoxetine induces CYP2C19 in cultured human hepatocytes, analogous to the macrolide antibiotics' effect on CYP3A4.
Consistent with the in vitro interaction with CYP2C19, fluoxetine has been reported to significantly decrease the in vivo clearance of CYP2C19 substrates diazepam (Lemberger et al., 1988) and S-mephenytoin (Jeppesen et al., 1996; Harvey and Preskorn, 2001). The major human metabolite of fluoxetine, norfluoxetine, may also be responsible for P450 inhibition, and this has not been addressed directly in the present studies; we have yet to examine whether norfluoxetine, which is equipotent against CYP2D6 (Stevens and Wrighton, 1993), shows time-dependent inhibition of CYP2C19 and 3A4. Moreover, because fluoxetine is a racemic mixture, clearly, there could be differences in the CYP3A4 and 2C19 interaction between the R and S form, as reported for CYP2D6 inhibition (Stevens and Wrighton, 1993). Fluoxetine has also been demonstrated to be a mechanism-based inhibitor of rat CYP2C11 (Murray and Murray, 2003).
While the potential of AZ1 to induce P450s in cultured human hepatocytes was being assessed, the compound was shown to cause significant loss of CYP2C9-mediated diclofenac 4′-hydroxylation, with minimal effect on CYP2C9 protein level as determined by Western blotting (data not shown). Prior to this, it had been established that AZ1 was not a potent time-dependent inhibitor of CYP2C9, using a recently described HLM assay (Atkinson et al., 2005). After further investigations at higher concentrations and longer time points than would have been used in a standard screen, AZ1 was identified as an extremely weak CYP2C9 inhibitor. The kinact/KI ratio (a measure of time-dependent inhibition efficiency) for AZ1, as determined in rCYP2C9, is 7 × 10–4 ml/min · nmol (KI 30 μM, kinact 0.02 min–1; Table 2), which is approximately 140 times smaller than that of tienilic acid (KI 2 μM, kinact 0.19 min–1, kinact/KI 0.1 ml/min ·nmol). Despite the relatively weak CYP2C9 time-dependent inhibition properties of AZ1, the inhibitory effect in cultured human hepatocytes was dramatic due to the suprapharmacological (free) compound concentrations used, the resultant “stoichiometry” of test compound to cells, and the long time course involved (typically 72 h with compound replenished every 24 h), which is exacerbated by low turnover of inhibitor, an increasingly common property for candidate drugs for oral therapies in which hepatic clearance is required to be low. Using a modified version of the physiological model developed by Ito et al. (2003) (eq. 4), P450 inhibition in cultured human hepatocytes can be quantitatively described. The original in vitro-in vivo model predicted accurately the 2-fold area under the plasma concentration-time curve increase of intravenously dosed midazolam following pretreatment with erythromycin (500 mg t.i.d.) (Ito et al., 2003) yet predicts minimal risk of significant CYP2C9-mediated clinical DDIs with a weak time-dependent inhibitor such as AZ1 (Fig. 6). However, the suppression of P450 activity in hepatocytes indicated the potential for reactive metabolite formation and prompted further investigations such as covalent binding work, leading to a better risk assessment of immune-related hypersensitivity reactions in the clinic.

Simulated profiles of P450 activity in human liver after oral dosing with AZ1 and erythromycin, respectively. Using the methodology of Ito et al. (2003), CYP2C9 activity in human liver was simulated after oral dosing with AZ1 (70 mg every 24 h for 72 h; open circles) and CYP3A4 activity in human liver after oral dosing with erythromycin (500 mg every 8 h for 72 h; closed circles). Physiological constants and the pharmacokinetic parameters of erythromycin needed for the simulation were taken directly from Ito et al. (2003) and are listed under Materials and Methods for AZ1.
This study has confirmed the importance of considering time-dependent P450 inhibition while using primary hepatocytes for assessing P450 induction, as cautioned previously (Silva and Nicoll-Griffith, 2002). The effect on P450 activity of a time-dependent inhibitor depends on the interplay between the efficiency of inactivation caused by parent (and/or subsequently formed metabolites) and the turnover rate of both parent and individual P450, together with any P450 induction. Clearly, reversible inhibitors can be removed by thoroughly washing cells before determination of P450 activity. As exemplified by AZ1, relatively weak time-dependent inhibitors that are cleared slowly may show dramatic inhibitory effects on P450 activity in primary culture over hours and/or days, especially since compound is traditionally replenished every 24 h. These data are particularly relevant because many oral drug candidates will be relatively metabolically stable, and some may demonstrate a degree of time-dependent P450 inhibition. Metabolically labile and/or potent time-dependent P450 inhibitors are likely to be eliminated from a lead-optimization screening cascade before entering resource-intensive P450 induction assays.
In conclusion, cultured human hepatocytes provide a sensitive system for studying time-dependent P450 inhibition. Together with a P450-selective substrate cassette approach, this provides an efficient assay that also flags the potential formation of reactive species and provides some indication of clinical toxicity risk. To fully evaluate the P450 induction potential of a drug candidate, mRNA and/or protein levels should be measured in addition to P450 activity, typically referred to as the “gold standard” endpoint, particularly if detailed knowledge of time-dependent P450 inhibition potential is lacking.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009969.
-
ABBREVIATIONS: P450, cytochrome P450; rP450, recombinant P450; CLint, intrinsic clearance; CLmet, metabolic clearance; fuinc, unbound fraction in incubation; fup, unbound fraction in plasma; [I], inhibitor concentration; KI, inhibition constant; kdeg, P450 degradation or turnover rate constant; kinact, maximal inactivation rate constant; DDI, drug-drug interaction; HMM, hepatocyte maintenance media; HLM, human liver microsome; DMSO, dimethyl sulfoxide; HPLC, high-performance liquid chromatography; MS/MS, tandem mass spectrometry.
- Received February 24, 2006.
- Accepted April 25, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}