


Abstract
Clopidogrel acyl-β-d-glucuronide is a mechanism-based inhibitor of cytochrome P450 2C8 in human liver microsomes (HLMs). However, time-dependent inactivation (TDI) of CYP2C8 could not be detected in an earlier study in human recombinant CYP2C8 (Supersomes). Here, we investigate whether different enzyme sources exhibit differences in detection of CYP2C8 TDI under identical experimental conditions. Inactivation of CYP2C8 by amiodarone (100 μM), clopidogrel acyl-β-d-glucuronide (100 μM), gemfibrozil 1-O-β-glucuronide (100 μM), and phenelzine (100 μM) was investigated in HLMs and three recombinant human CYP2C8 preparations (Supersomes, Bactosomes, and EasyCYP Bactosomes) using amodiaquine N-deethylation as the marker reaction. Furthermore, the inactivation kinetics of CYP2C8 by clopidogrel glucuronide (5–250 μM) was determined in Supersomes and Bactosomes. Amiodarone caused weak TDI in all enzyme preparations tested, while the extent of inactivation by clopidogrel glucuronide, gemfibrozil glucuronide, and phenelzine varied markedly between preparations, and even different Supersome lots. Both glucuronides caused strong inactivation of CYP2C8 in HLMs, Bactosomes and in one Supersome lot (>50% inhibition), but significant inactivation could not be reliably detected in other Supersome lots or EasyCYP Bactosomes. In Bactosomes, the concentration producing half of kinact (KI) and maximal inactivation rate (kinact) of clopidogrel glucuronide (14 μM and 0.054 minute−1) were similar to those determined previously in HLMs. Phenelzine caused strong inactivation of CYP2C8 in one Supersome lot (91% inhibition) but not in HLMs or other recombinant CYP2C8 preparations. In conclusion, different enzyme sources and different lots of the same recombinant enzyme preparation are not equally sensitive to detect inactivation of CYP2C8, suggesting that recombinant CYPs should be avoided when identifying mechanism-based inhibitors.
Introduction
Cytochrome P450 2C8 (CYP2C8) is recognized as an important drug-metabolizing enzyme, and inhibition of CYP2C8 can cause many clinically significant drug-drug interactions (Backman et al., 2016). Mechanism-based inhibition (MBI) of CYP2C8 is of particular importance, since it inactivates the enzyme irreversibly or quasi-irreversibly, thus causing long-lasting loss of catalytic activity. MBI requires a catalytic transformation of the inactivator to a reactive intermediate or metabolite that inactivates the enzyme, and the inactivation process is thus time-dependent (Silverman, 1995). Established examples of clinically significant, strong mechanism-based inhibitors of CYP2C8 are the acyl glucuronide metabolites of the lipid-lowering drug gemfibrozil and the antiplatelet agent clopidogrel. These glucuronides are formed by UGT2B enzymes from their parent compounds and have been shown to cause MBI of CYP2C8 in human liver microsomes (HLMs) (Ogilvie et al., 2006; Mano et al., 2007; Tornio et al., 2014; Kahma et al., 2018). However, in a previous study investigating the interaction between cerivastatin and clopidogrel, time-dependent inactivation (TDI) of CYP2C8 by clopidogrel acyl-β-d-glucuronide was not observed when incubated with human recombinant CYP2C8 expressed in baculovirus-infected insect cells (Supersomes) (Floyd et al., 2012). Likewise, in preliminary studies, we were also unable to detect inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide in Supersomes.
Recombinantly expressed human cytochrome P450 (P450) enzymes are widely available and commonly used for screening metabolic drug-drug interactions to identify inhibitors of P450 enzymes (Parkinson et al., 2010). Several studies report large differences in kinetic and inhibition parameters between different enzyme sources, but little is known about the effects of different enzyme sources on mechanism-based inhibition (Kumar et al., 2006; Van et al., 2007; Parkinson et al., 2010). Reliable in vitro identification of P450 inhibitors is crucial to detecting possible clinically significant drug-drug interactions of compounds already in the preclinical phase. This is especially important for mechanism-based inhibitors, since irreversible P450 inactivation caused by mechanism-based inhibition can pose greater safety concerns compared with reversible inhibition. To investigate the sensitivity and reliability of different enzyme sources in mechanism-based inhibition of CYP2C8, we studied the performance of HLMs and three different recombinant human P450 preparations under controlled experimental conditions using clopidogrel acyl-β-d-glucuronide, gemfibrozil 1-O-β-glucuronide, amiodarone, phenelzine, and raloxifene as CYP2C8 inhibitors (Supplemental Table 1).
Materials and Methods
Materials.
Pooled HLMs (UltraPool, lots 38290 and 38291) and human recombinant CYP2C8 expressed in baculovirus-infected insect cells with human cytochrome b5 (Supersomes, lots 03459, 32804, 3221713, 5057003 and 7053001) were purchased from Corning (Corning, NY). Human recombinant CYP2C8 enzymes expressed in Escherichia coli with human cytochrome b5 (low reductase Bactosomes, lot C2C8BLR001, and EasyCYP Bactosomes, lot C2C8BLR002) and membrane protein isolated from E. coli containing empty expression plasmid (control Bactosomes, lot BAC092) were from Cypex (Dundee, UK). The rapid equilibrium dialysis device (RED) was purchased from Thermo Scientific (Waltham, MA). Amiodarone hydrochloride, amodiaquine dihydrochloride dehydrate and β-NADPH were from Sigma-Aldrich (St. Louis, MO). Clopidogrel acyl-β-d-glucuronide was purchased from Santa Cruz Biotechnology (Dallas, TX). Gemfibrozil 1-O-β-glucuronide, N-desethylamodiaquine hydrochloride, N-desethylamodiaquine-d5, phenelzine sulfate and raloxifene hydrochloride were from Toronto Research Chemicals (Toronto, ON, Canada). Bovine serum albumin (BSA) was purchased from Biowest (Nuaillé, France). Other solvents and reagents were of analytical grade.
Incubation Conditions.
Time-dependent inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide (5–250 μM), gemfibrozil 1-O-β-glucuronide (100 μM), amiodarone (100 μM), phenelzine (100 μM), and raloxifene (1 μM) was studied in HLMs, Supersomes, EasyCYP Bactosomes, and low-reductase Bactosomes using amodiaquine N-deethylation as the marker reaction for CYP2C8 activity. Incubations were conducted in 96-well plates using Tecan Freedom EVO 150 automated liquid handler (Männedorf, Switzerland). Inhibitors diluted in 0.1 M sodium phosphate buffer (pH 7.4) were prewarmed on a heated shaker (37°C, 350 rpm) for 3 minutes with microsomes (0.5 mg/ml), followed by addition of NADPH (1 mM) and preincubation for 0, 2.5, 6, 12, 20, and 30 minutes. To minimize competitive CYP2C8 inhibition during the activity phase, protein and inhibitor concentrations were then diluted 20-fold by removing aliquots of the preincubation mixture to wells containing amodiaquine (10 μM) and NADPH (1 mM) in buffer. The reaction mixture was incubated for 2 minutes (37°C, 350 rpm) and was stopped by mixing one part sample and three parts cold (4°C) stop solution (0.3% formic acid in acetonitrile/methanol/water 90:5:5 v/v/v) containing deuterium-labeled N-desethylamodiaquine as an internal standard. Samples were centrifuged for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Incubations were performed in at least triplicates.
Clopidogrel acyl-β-d-glucuronide and phenelzine were dissolved in water, gemfibrozil 1-O-β-glucuronide, and amiodarone in methanol and raloxifene in ethanol/water (4:1 v/v). The final solvent concentrations in incubations were 1% or less. The nonspecific binding of 100 μM clopidogrel acyl-β-d-glucuronide to HLMs, CYP2C8 Supersomes (lots 03459, 3221713 and 7053001), low reductase CYP2C8 Bactosomes and low reductase CYP2C8 EasyCYP Bactosomes (0.5 mg/ml) was determined by equilibrium analysis using the rapid equilibrium dialysis (RED) system.
Quantification of Drug Concentrations.
N-Desethylamodiaquine concentrations were determined using a Shimadzu Nexera liquid chromatography system coupled to a Shimadzu 8050 tandem mass spectrometer (Shimadzu Corporation, Kyoto, Japan). The analytical method was modified from a previously published method (Filppula et al., 2012). The only changes were a smaller injection volume (1 μl) and faster gradient (5%–100% B in 8 minutes). The lower limit of quantification of N-desethylamodiaquine was 0.001 μM and the upper limit of quantification was 3.125 μM.
Clopidogrel, clopidogrel acyl-β-d-glucuronide, gemfibrozil, and gemfibrozil-1-O-β-glucuronide concentrations were measured with a Nexera X2 liquid chromatograph (Shimadzu) coupled to a 5500 Qtrap or API3000 tandem mass spectrometer (AB Sciex, Toronto, ON, Canada). Clopidogrel and clopidogrel acyl-β-d-glucuronide were separated on a Sun Fire C18 analytical column (100 × 2.1 mm i.d., 3-μm particle size; Waters, Milford, MA) using 2 mM ammonium acetate (pH 5.5) (mobile phase A) and acetonitrile (mobile phase B). The proportion of mobile phase B was linearly increased from 20% to 55% over 2.8 minutes, then maintained at 90% for 1.2 minutes and equilibrated back to 20%. The mobile phase flow rate and column oven temperature were set at 0.26 ml/min and 30°C. The mass spectrometer was operated in positive polarity multireaction mode (MRM+) utilizing the target mass-to-charge transitions (m/z) of 322 to 155 and 484 to 308 for clopidogrel and clopidogrel acyl-β-d-glucuronide. Deuterium-labeled clopidogrel and deuterium-labeled clopidogrel carboxylic acid served as internal standards.
The separation of gemfibrozil and gemfibrozil-1-O-β-glucuronide was performed on a Kinetex C18 column (75 × 2.1 mm i.d., 2.6-μm particle size; Phenomenex, Torrance, CA) using 2 mM ammonium acetate (pH 4) and acetonitrile (mobile phase B) in following gradient conditions: 1 minute at 20% B on hold, then a linear increase from 20% B to 90% B over 4 minutes followed by equilibration back to the starting composition (20% B). The mass spectrometric detection of gemfibrozil and gemfibrozil-1-O-β-glucuronide used as a basis their characteristic negative mode (MRM-) transitions of 249 to 121 and 425 to 121, including the corresponding deuterium-labeled internal standards. The lower limits of quantitation of clopidogrel, clopidogrel acyl-β-d-glucuronide, gemfibrozil, and gemfibrozil-1-O-β-glucuronide were 1, 0.5, 1 and 0.5 μM, respectively, and the day-to-day coefficient of variation were below 15% at relevant concentrations for all analytes.
Effects of BSA and Microsomal Protein on CYP2C8 Inactivation by Clopidogrel Acyl-β-D-Glucuronide.
The effects of fatty acid binding by albumin on CYP2C8 inactivation by clopidogrel acyl-β-d-glucuronide were investigated in Supersomes. CYP2C8 Supersomes (0.5 mg/ml), clopidogrel acyl-β-d-glucuronide (100 μM) and NADPH (1 mM) were preincubated with BSA (0.1% w/v), followed by dilution and incubation with amodiaquine (10 μM) as described in incubation conditions.
To investigate the effect of microsomal protein on CYP2C8 inactivation, low reductase Bactosomes were diluted with control Bactosomes isolated from E. coli host strain and sodium phosphate buffer to give similar CYP2C8 and microsomal protein concentrations as in EasyCYP Bactosomes (Table 1). Time-dependent inactivation of CYP2C8 by 100 μM clopidogrel acyl-β-d-glucuronide was studied in diluted Bactosomes as described in incubation conditions.
Concentrations of CYP2C8 and other constituents in preincubations in the different enzyme preparations
Metabolic Depletion of Clopidogrel Acyl-β-D-Glucuronide and Gemfibrozil 1-O-β-Glucuronide.
The extent of metabolic depletion of clopidogrel acyl-β-d-glucuronide (100 μM) and gemfibrozil 1-O-β-glucuronide (100 μM) during preincubation was studied in HLMs and CYP2C8 Supersomes. Following a 3-minute preincubation without NADPH (as described earlier), the inhibitor was preincubated for 0, 10 and 30 minutes with microsomes (0.5 mg/ml) and NADPH (1 mM) in 0.1 M sodium phosphate buffer (pH 7.4) with and without the β-glucuronidase inhibitor saccharolactone (5 mM). The reaction was stopped by mixing one part sample and three parts cold (4°C) acetonitrile and formic acid (99:1 v/v) containing either clopidogrel-d4 and clopidogrel-d4 carboxylic acid or gemfibrozil-d6 and gemfibrozil-d6 1-O-β-glucuronide as internal standards for clopidogrel and gemfibrozil LC-MS/MS methods, respectively.
The Effect of Clopidogrel on the CYP2C8 Inactivation Potency of Clopidogrel Acyl-β-D-Glucuronide in HLMs.
The effect of parent clopidogrel in the time-dependent inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide was studied by preincubating 4, 40, or 200 μM clopidogrel (and control containing no clopidogrel) with clopidogrel acyl-β-d-glucuronide (100 μM), HLMs (0.5 mg/ml) and NADPH (1 mM), followed by dilution and incubation, as described in incubation conditions.
Determination of Kinetic Parameters of Inactivation.
The maximal inactivation rate (kinact) and the inhibitor concentration needed to cause half of kinact (KI) were determined for clopidogrel acyl-β-d-glucuronide in Supersomes and Bactosomes. Various inhibitor concentrations were incubated as described above and based on these results, kinact and KI were estimated by nonlinear regression in SigmaPlot, version 13.0 (Systat Software, San Jose, CA) using the following equation: (1)where kobs is the initial inactivation rate constant of CYP2C8 inactivation by each inhibitor concentration [I].
(1)where kobs is the initial inactivation rate constant of CYP2C8 inactivation by each inhibitor concentration [I].
Spectral Binding.
The binding affinity of clopidogrel acyl-β-d-glucuronide to CYP2C8 in Supersomes and Bactosomes was studied in spectral binding experiments. CO difference spectral scanning was first performed to determine the CYP2C8 concentration in Bactosomes. Supersomes or Bactosomes were then diluted in 0.1 M potassium phosphate buffer (pH 7.4) to a final CYP2C8 concentration of 0.5 μM. Aliquots (400 μl each) of diluted CYP2C8 were placed into two quartz cuvettes. The cuvettes were placed in the spectrophotometer and allowed to equilibrate to 25°C for 10 minutes, after which a baseline spectrum was obtained. Clopidogrel acyl-β-d-glucuronide was titrated (1 μl additions) into the sample cuvette over a range of concentrations from 0.12 to 12 μM, using a positive displacement pipette. With each addition of ligand to the sample cuvette, 1 μl of 4:1 MeOH/DMSO was added to the reference cuvette. After each titration, a difference spectrum was recorded. Spectra were recorded on a Modernized Aminco DW-2 UV/Vis spectrophotometer controlled with Olis Spectral Works software v. 5.888.181 (Olis Inc, Bogart, GA). The cuvette temperature was held constant at 25°C with an external water bath temperature controller. The spectrophotometer was set to scan from 500 to 350 nm and average six data points at every 0.5 nm interval. The baseline spectrum was subtracted individually from each titration spectra. Each spectra was normalized to P450 content. The difference in absorbance between the peak (Amax) and trough (Amin) was determined for each titration (Supersomes: A406–A425; Bactosomes: A405–A422) and plotted as a function of clopidogrel acyl-β-d-glucuronide concentration. An Eadie-Hofstee transformation was used to determine linearity indicating a single binding event. Using GraphPad Prism, version 6.07 (GraphPad Software, La Jolla, CA), a nonlinear regression analysis was performed to determine apparent spectral dissociation rate constants (Ks) of clopidogrel acyl-β-d-glucuronide to CYP2C8 Supersomes and CYP2C8 Bactosomes (Locuson et al., 2007).
Results
Nonspecific Binding of Clopidogrel Acyl-β-D-Glucuronide.
The unbound fractions of 100 μM clopidogrel acyl-β-d-glucuronide in HLMs, CYP2C8 Supersomes (lots 03459, 3221713 and 7053001), CYP2C8 Bactosomes and CYP2C8 EasyCYP Bactosomes varied from 0.90 to 0.95, indicating only 5%–10% nonspecific binding (Table 1).
Time-Dependent Inactivation of CYP2C8 by Amiodarone, Clopidogrel Acyl-β-D-Glucuronide, Gemfibrozil 1-O-β-Glucuronide, Phenelzine, and Raloxifene.
The variability between enzyme preparations in TDI of CYP2C8 was small for amiodarone. Preincubation of amiodarone (100 μM) with NADPH for 30 minutes caused 47%–55% inhibition of CYP2C8 activity in HLMs, Bactosomes and Supersome lot 7053001 (Fig. 1A). A slightly weaker inactivation of CYP2C8 was observed in EasyCYP Bactosomes and Supersome lot 5057003 (34%–36% inhibition).

Time-dependent inactivation of CYP2C8 by 100 μM amiodarone (A), 100 μM clopidogrel acyl-β-D-glucuronide (B), 100 μM gemfibrozil 1-O-β-glucuronide (C), and 100 μM phenelzine (D) in HLMs, Bactosomes, EasyCYP Bactosomes, and Supersome lots 5057003 and 7053001. Each point represents the mean ± S.D. of quadruplicate incubations.
Considerably higher variation in TDI results was observed for clopidogrel acyl-β-d-glucuronide, gemfibrozil 1-O-β-glucuronide and phenelzine (Fig, 1B-D). After 30 minutes, 100 μM clopidogrel acyl-β-d-glucuronide caused 72% inhibition of CYP2C8 activity in HLMs, 63% inhibition in Bactosomes and 54% inhibition in Supersome lot 7053001, but significant inactivation could not be reliably detected in three other Supersome lots (03459, 3221713 and 5057003) or EasyCYP Bactosomes (Figs. 1 and 2). In CYP2C8 Bactosomes mixed with control Bactosomes, clopidogrel acyl-β-d-glucuronide produced only a slight reduction in CYP2C8 activity (22% inhibition), comparable to that in EasyCYP Bactosomes (Fig. 1B).

The initial inactivation constants (kobs) (minute−1) of 100 μM amiodarone (AMD), 100 μM clopidogrel acyl-β-d-glucuronide (CAG), 100 μM gemfibrozil 1-O-β-glucuronide (GFG), 100 μM phenelzine (PHZ), and 1 μM raloxifene (RLX) in different enzyme sources.
A similar trend was observed for gemfibrozil 1-O-β-glucuronide. After 30 minutes, CYP2C8 inactivation by 100 μM gemfibrozil 1-O-β-glucuronide was prominent in Supersome lot 7053001 (95% inhibition), HLMs (91% inhibition), and Bactosomes (88% inhibition) (Fig. 1C). In EasyCYP Bactosomes and Supersome lot 5057003, inactivation was considerably slower and weaker, reaching 72% and 66% inhibition at 30 minutes, respectively.
Preincubation with 100 μM phenelzine, on the other hand, resulted in significant TDI only in Supersome lot 7053001 (91% inhibition after 30 minutes), which had the highest CYP2C8 activity, while TDI in HLMs, Bactosomes, EasyCYP Bactosomes and another Supersome lot was only 35% or less. To rule out high CYP2C8 activity as the cause for the rapid inactivation in Supersome lot 7053001, we preincubated 100 μM phenelzine with NADPH in diluted Supersomes (lot 7053001, 0.1 mg/ml), and found that dilution of Supersomes did not affect inactivation of CYP2C8 by phenelzine (89% inhibition at 30 minutes) (Fig. 1D).
Raloxifene (1 μM) caused only slight TDI in Bactosomes, EasyCYP Bactosomes and Supersome lot 7053001 (18%–20% inhibition after 30 minutes, data not shown). No relevant CYP2C8 inactivation by raloxifene could be detected in HLMs or Supersome lot 5057003 (inhibition 10% or less).
Effect of BSA on the Inactivation of CYP2C8 by Clopidogrel Acyl-β-d-Glucuronide.
The effect of BSA was studied in the Supersome lots with the weakest CYP2C8 inactivation (lots 03459, 3221713, and 5057003). Addition of 0.1% w/v BSA slightly reduced the unbound fraction of clopidogrel acyl-β-d-glucuronide (Table 1), and did not have a significant effect on CYP2C8 inactivation in any of the Supersome lots tested (Figs. 2 and 3).

Residual CYP2C8 activity after preincubating 100 μM clopidogrel acyl-β-d-glucuronide for 0 and 30 minutes in Supersome lots 03459, 3221713, 5057003, and 7053001 with and without BSA. Each bar represents the mean ± S.D. of quadruplicate incubations.
Metabolic Depletion of Clopidogrel Acyl-β-d-Glucuronide and Gemfibrozil 1-O-β-Glucuronide.
No significant depletion of clopidogrel acyl-β-d-glucuronide or gemfibrozil 1-O-β-glucuronide could be observed in Supersomes or HLMs during a 30-minute preincubation with NADPH (Fig. 4). However, some loss of clopidogrel acyl-β-d-glucuronide had occurred in Supersomes already during the 3-minute equilibration at 37°C prior to NADPH addition initiating the preincubation, which was apparently owing to conversion to clopidogrel. Accordingly, at the start of preincubation, clopidogrel concentration was higher in Supersome samples (7 and 6 μM at 0 and 30 minutes, respectively) than in HLM samples (3 and <1 μM at 0 and 30 minutes, respectively). Gemfibrozil concentration was similar in all samples (<1 μM). However, the β-glucuronidase inhibitor saccharolactone had no effect on the concentrations of clopidogrel or gemfibrozil or the depletion of clopidogrel acyl-β-d-glucuronide or gemfibrozil 1-O-β-glucuronide in the incubations.

Depletion of 100 μM clopidogrel acyl-β-d-glucuronide (A) and 100 μM gemfibrozil 1-O-β-glucuronide (B) in HLMs and Supersomes with and without saccharolactone. Each point represents the mean ± S.D. of triplicate incubations.
Effect of Clopidogrel on the CYP2C8 Inactivation Potency of Clopidogrel Acyl-β-D-Glucuronide in HLMs.
Addition of 4 or 40 μM of clopidogrel to the 30-minute preincubation in HLMs did not have a significant effect on the inactivation rate of CYP2C8 by clopidogrel acyl-β-d-glucuronide, and addition of 200 μM clopidogrel decreased TDI only slightly (61% and 66% inhibition at 30 minutes, with and without clopidogrel, respectively). The kobs values of CYP2C8 inactivation by 100 μM clopidogrel acyl-β-d-glucuronide with 0, 4, 40, and 200 μM clopidogrel were 0.046, 0.038, 0.039, and 0.027 minute−1, respectively (Fig. 2).
CYP2C8 Inactivation Kinetics in Supersomes and Bactosomes.
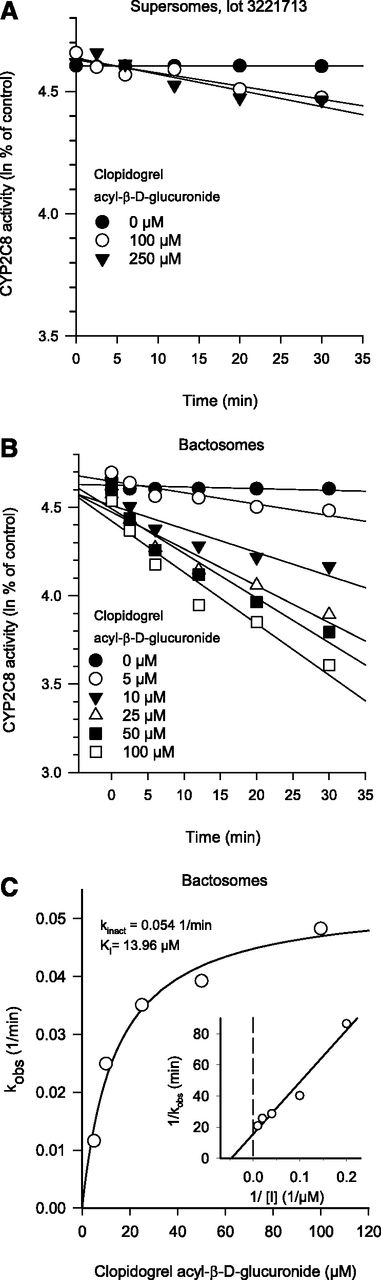
In inactivation kinetic experiments with Supersome lot 3221713, inactivation could not be detected even with 250 μM clopidogrel acyl-β-d-glucuronide (Fig. 5A). In Bactosomes, inhibition of CYP2C8 activity by clopidogrel acyl-β-d-glucuronide was concentration- and time-dependent, with KI and kinact of 14 μM and 0.054 minute−1, respectively (Fig. 5, B and C).

Time and concentration-dependent inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide in Supersome lot 3221713 (A) and low reductase Bactosomes (B) and respective kobs vs. concentration plot (C). Each point represents the mean ± S.D. of quadruplicate incubations.
Binding of Clopidogrel Acyl-β-D-Glucuronide to Supersomes and Bactosomes.
Spectral binding studies were conducted to investigate whether the inhibitor affinity of clopidogrel acyl-β-d-glucuronide to CYP2C8 differed between Supersomes and Bactosomes. The spectral dissociation constant, Ks, was lower in Bactosomes (0.41 ± 0.06 μM) compared with Supersomes (1.4 ± 0.2 μM, lot 5057003) (Fig. 6; Supplemental Fig. 1).

The difference in absorbance (B) between the peak (Amax) and trough (Amin) observed with increasing concentrations of clopidogrel acyl-β-glucuronide, using CYP2C8 Bactosomes (A) and two different lots of CYP2C8 Supersomes (B and C). The data were transformed to plot the absorbance difference as a function of the ratio of the absorbance difference and substrate concentration (D–F). Once linearity was determined, the original plots were fitted to a hyperbolic function to determine the apparent spectral dissociation rate constants (Ks) of clopidogrel acyl-β-glucuronide to CYP2C8 enzyme systems.
Discussion
The results of the present study show that inactivation of CYP2C8 by four known mechanism-based CYP2C8 inhibitors varies considerably between different enzyme preparations. Whereas inactivation of CYP2C8 was weak or almost absent in some of the recombinant enzyme preparations, particularly certain Supersome lots, inactivation of CYP2C8 by the clinically significant inhibitors (glucuronides of clopidogrel and gemfibrozil) could generally be reliably detected in HLMs. Moreover, the degree of variation between the preparations was to some extent inhibitor-dependent. Clopidogrel acyl-β-d-glucuronide and gemfibrozil 1-O-β-glucuronide caused strong inactivation in HLMs, Bactosomes, and Supersome lot 7053001 but had little or only moderate effect in the other Supersome lots and EasyCYP Bactosomes. The kinetic parameters of inactivation of clopidogrel acyl-β-d-glucuronide determined in Bactosomes were similar to those determined previously in HLMs (KI 9.9 μM and kinact 0.047 minute−1) (Tornio et al., 2014). In contrast, phenelzine caused strong inactivation only in Supersome lot 7053001, and considerably weaker inactivation in the other enzyme preparations, including HLMs, which resembles the previously reported 45-fold smaller KI of phenelzine in recombinant CYP2C8 than in HLMs (Polasek et al., 2004) (Supplemental Table 2).
A similar discrepancy in CYP2C8 inactivation between recombinant CYP2C8 and HLMs has been previously reported also for amiodarone (Polasek et al., 2004) (Supplemental Table 2). In our experiments, however, CYP2C8 inactivation by amiodarone (100 μM) was weak and showed only little variation between enzyme sources (34%–55% inhibition after 30 minutes). On the other hand, CYP2C8 inactivation by 1 μM raloxifene was substantially weaker in all preparations compared with the previously reported inactivation parameters in Supersomes (KI 0.26 μM and kinact 0.1 minute−1) (VandenBrink et al., 2012). Notably, in contrast to gemfibrozil and clopidogrel, phenelzine, amiodarone, and raloxifene have not been shown to cause clinically relevant CYP2C8 inhibition.
To examine the potential causes for the observed variation in CYP2C8 inactivation potency, we performed a range of experiments. Spectral binding data suggests that clopidogrel acyl-β-d-glucuronide binds more tightly to CYP2C8 in Bactosomes than in Supersomes, which might partially explain the lack of significant inactivation in certain Supersome lots. According to the manufacturers, the primary sequences of the recombinant CYP2C8 were identical to the native sequence of human CYP2C8, with the exception of alanine and proline added to the N-terminal end in Bactosomes. Thus, differences in the primary protein sequence are unlikely to explain the observed differences in binding and inactivation potency. Yet, functionality of the enzyme can be altered also by differences in cellular environment between human, insect, and bacterial cells, such as different lipid composition of the membrane, pH, oxidative stress, and chaperone proteins (Marheineke et al., 1998; Nannenga and Baneyx, 2011; Brignac-Huber et al., 2016). These differences could affect the conformation, folding, or positioning of P450s in membranes, which in turn could prevent covalent modifications or metabolite-intermediate complexation caused by mechanism-based inhibitors. It has been proposed that positioning of P450 enzymes on the insect cell membranes might differ from their positioning in the membrane of the endoplasmic reticulum in human hepatocytes (Zhang et al., 2004). Moreover, membrane-bound CYP2C8 has been shown to form homomeric dimers, which can cause conformational changes in the F/G loops of the enzymes, possibly affecting substrate binding and inhibiting metabolism of hydrophobic substrates (Schoch et al., 2004; Hu et al., 2010; Reed and Backes, 2017). The aggregation properties of P450s are affected by the microsomal lipid composition, the lipid/protein ratio, and the concentrations of the redox partners of P450 enzymes (Backes and Kelley, 2003), which can differ markedly between expression systems and human cells. In addition, the lipid composition of the membrane affects the interaction of P450s and their redox partners (Brignac-Huber et al., 2016). Interestingly, the differences in strength of inhibition between the tested preparations showed a trend for correlation with cytochrome c reductase activity but not with the other variables (Supplemental Fig. 2; Table 1).
Theoretically, the presence of relatively higher concentrations of microsomal protein and possibly different lipid composition in EasyCYP Bactosomes and some of the Supersome lots could result in greater nonspecific binding of the inhibitors, reducing their apparent inhibitory effects. To reduce these possibilities, we used the same total protein concentration in all experiments. Accordingly, the fraction unbound of clopidogrel acyl-β-d-glucuronide was similar (0.90–0.92) in all the recombinant enzyme preincubation mixtures. However, when we added control Bactosome membrane protein to low reductase Bactosomes to get a similar composition as in EasyCYPs, clopidogrel acyl-β-d-glucuronide produced a likewise slow reduction in CYP2C8 activity as in EasyCYP Bactosomes. Thus, the addition of microsomal constituents weakens the sensitivity to detect TDI of CYP2C8 by clopidogrel acyl-β-d-glucuronide in Bactosomes. As microsomal preparations contain long-chain fatty acids that markedly inhibit CYP2C8 (Palacharla et al., 2017), we tested whether sequestration of fatty acids with albumin increases the CYP2C8 inhibitory effect of clopidogrel acyl-β-d-glucuronide in Supersomes. However, addition of 0.1% w/v BSA did not augment the inhibition caused by clopidogrel acyl-β-d-glucuronide.
Finally, variation in the depletion of inhibitor during preincubation may also contribute to differences in the degree of inactivation between different enzyme sources (Van et al., 2007). Glucuronide hydrolysis by β-glucuronidase has been observed previously in HLMs and insect cell preparations (Oleson and Court, 2008). In our experiments, depletion of clopidogrel and gemfibrozil glucuronides was negligible during the 30-minute preincubation in HLMs and Supersomes and was not influenced by the β-glucuronidase inhibitor saccharolactone. However, about 1/10 of clopidogrel acyl-β-d-glucuronide had been converted to clopidogrel during the 3-minute equilibration before the start of the 30-minute preincubations in Supersomes, whereas only a small amount of clopidogrel had been formed in HLMs. It is noteworthy that in the presence of methanol, clopidogrel acyl-β-d-glucuronide can be converted to clopidogrel (Silvestro et al., 2011), and therefore we avoided the use of methanol as a solvent. As clopidogrel acyl-β-d-glucuronide was stable during the preincubation and not influenced by saccharolactone, the reason for formation of clopidogrel before the preincubation remains unknown. Nevertheless, the concentration of clopidogrel during the preincubation (6 to 7 μM) could have been high enough to cause competitive inhibition of CYP2C8, since its half-maximal inhibitory concentration (IC50) ranges from 2.8 to 53.6 μM in vitro (Walsky et al., 2005; Floyd et al., 2012; Tornio et al., 2014). Such competitive inhibition could reduce TDI of CYP2C8. However, addition of up to 200 μM clopidogrel to a standard preincubation in HLMs resulted only in minor decreases of CYP2C8 inactivation by 100 μM clopidogrel acyl-β-D-glucuronide.
While gemfibrozil 1-O-β-glucuronide, clopidogrel acyl-β-d-glucuronide, and amiodarone are considered to be mechanism-based inhibitors of CYP2C8, CYP2C8 inhibition by phenelzine is not fully consistent with mechanism-based inhibition and is at least partly reversible (Polasek et al., 2004; Ogilvie et al., 2006; Tornio et al., 2014). Mechanistic differences may partially explain why we were unable to show inactivation of CYP2C8 by phenelzine in most experiments, including those with HLMs, whereas phenelzine caused a particularly strong inactivation in the CYP2C8 Supersome lot that was most sensitive to the other inhibitors. Of note, similar disparities have been observed also with other monoamine inhibitors, tricyclic antidepressants, cimetidine, and tamoxifen (Supplemental Table 2) (Zhao et al., 2002; Madeira et al., 2004; Polasek et al., 2004; Polasek and Miners, 2008), where TDI has usually been considerably stronger in recombinant enzyme systems than in HLMs or hepatocytes. Moreover, in one study with MDMA, CYP2D6 Bactosomes produced significantly higher KI values compared with CYP2D6 Supersomes and yeast microsomes (Van et al., 2007). Unlike amiodarone and phenelzine, the glucuronides of gemfibrozil and clopidogrel have been shown to cause CYP2C8 inactivation at clinically relevant doses of the parent drugs, leading to clinically important drug-drug interactions (Backman et al., 2002, 2016; Niemi et al., 2003; Itkonen et al. 2016, 2018, 2019a, 2019b; Jaakkola et al., 2005; Karonen et al., 2010; Filppula et al., 2013; Tornio et al., 2014; Menon et al., 2015). Their CYP2C8 inactivating effect was strong in HLMs, suggesting that HLMs are the most reliable system to screen clinically relevant inactivation of CYP2C8. We are not aware of any clinically relevant mechanism-based inhibitors of P450 enzymes whose TDI effect could not be observed in HLMs.
Taken together, our results indicate that there are major differences between enzyme sources in sensitivity to detect TDI of CYP2C8. The differences are inhibitor-dependent, which makes the phenomenon complex. Whereas TDI of CYP2C8 by the glucuronides of clopidogrel and gemfibrozil was strong in HLMs and Bactosomes and extremely weak or even absent in some Supersome lots, TDI by the nonglucuronide compound phenelzine could only be observed in Supersomes. Our findings suggest that neither nonspecific binding, depletion of inhibitor, nor inhibitory fatty acids explain the weak inactivation in some enzyme preparations. However, differences in other microsomal constituents, binding affinity, and conversion of clopidogrel acyl-β-d-glucuronide to clopidogrel may play some role. Generally, it appears that even slight differences between lots of recombinant enzymes may lead to inability to detect mechanism-based inhibition. Further studies are needed to elucidate the causes of such variation. Nonetheless, it is important to recognize that although human liver microsomes are generally reliable and produce clinically useful results, there is a risk of false negatives if recombinant enzymes are used to detect mechanism-based inhibition of P450 enzymes.
Authorship Contributions
Participated in research design: Kahma, Filppula, Totah, Backman.
Conducted experiments: Kahma, Evangelista, Launiainen, Viinamäki, Neuvonen.
Performed data analysis: Kahma, Evangelista, Backman.
Wrote or contributed to the writing of the manuscript: All authors.
Footnotes
- Received November 13, 2018.
- Accepted January 24, 2019.
This study has been financially supported by a grant from the Academy of Finland [Grant decision 278123, 2014] and by State funding for university-level health research, Finland.
This work was previously presented as a poster at the 1st Nordic Conference on Personalized Medicine (NORPM), May 30 – June 1, 2018, Nyborg, Denmark.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BSA
- bovine serum albumin
- HLMs
- human liver microsomes
- KI
- inhibitor concentration that produces half-maximal rate of inactivation
- kinact
- maximal inactivation rate constant
- kobs
- initial inactivation rate constant
- Ks
- dissociation constant
- MBI
- mechanism-based inhibition
- MDMA
- 3,4-methylenedioxy methamphetamine
- P450
- cytochrome P450
- TDI
- time-dependent inactivation
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}