


Visual Overview

Abstract
Piperine (PPR) is the representative alkaloid component of the piper species (family: Piperaceae). Our rapid screening study found PPR caused time-dependent inhibition of cytochrome P450s (CYP) 3A and 2D6, and CYP3A was inactivated the most. Further study demonstrated that PPR is a time-, concentration-, and NADPH-dependent inhibitor of CYP3A, and significant loss (49.5% ± 3.9%) of CYP3A activity was observed after 20minute incubations with 80 μM PPR at 37°C. The values of KI and kinact were 30.7 μM and 0.041 minutes−1, respectively. CYP3A competitive inhibitor ketoconazole showed protective effect against the enzyme inactivation. Superoxide dismutase/catalase and GSH displayed minor protection against the PPR-caused enzyme inactivation. Ferricyanide partially reduced the enzyme inhibition by PPR. Additionally, NADPH-dependent formation of reactive metabolites from PPR were found in human liver microsomal incubation mixtures. An ortho-quinone intermediate was trapped by NAC in microsomal incubations with PPR. DM-PPR, demethylene metabolite of PPR, showed weak enzyme inactivation relative to that caused by PPR. It appears that both carbene and ortho-quinone intermediates were involved in the inactivation of CYP3A caused by PPR.
SIGNIFICANCE STATEMENT CYP3A subfamily members (mainly CYP3A4 and CYP3A5) play a critical role in drug metabolism. Piperine (PPR), a methylenedioxyphenyl derivative combined with an unsaturated ketone, is the major active ingredient of pepper. PPR revealed time-, concentration-, and NADPH-dependent inhibitory effect on CYP3A. Carbene and quinone metabolites were both involved in the observed CYP3A inactivation by PPR. Apparently, the unsaturated ketone moiety did not participate in the enzyme inactivation. The present study sounds an alert of potential risk for food-drug interactions.
Introduction
Piperis fructus, nearly maturely or maturely dried fruits of Piper nigrum, are commonly used as dietary spices, such as food additives and condiments (Wattanathorn et al., 2008). Piperine (1-piperoylpiperidine, PPR) is a dominant alkaloid occurring in the fruits of long pepper (Piper longum Linn), black pepper (Piper nigrum Linn), and other piper species (family: Piperaceae) (Wattanathorn et al., 2008; Umar et al., 2013; Gupta et al., 2015). In addition, pepper has had a long history of medicinal use in Asian and Pacific islands (Bae et al., 2010; Umar et al., 2013). It is also often employed to treat diseases related to the gastrointestinal tract, particularly in Ayurvedic medicines (Johri and Zutshi, 1992; Gupta et al., 2015). PPR was reported to possesses multiple excellent and extensive pharmacological effects, such as anti-inflammatory (Bang et al., 2009; Murunikkara et al., 2012; Tasleem et al., 2014), analgesic (Tasleem et al., 2014), antioxidant (Chonpathompikunlert et al., 2010; Zarai et al., 2013; Samra et al., 2016), bioavailability enhancer (Katiyar et al., 2016), and anticonvulsant effect (Bukhari et al., 2013). Additionally, PPR reportedly attenuated collagenase-mediated tendon injury (Gong et al., 2017). The structure of PPR can be divided into three parts: heteroaromatic ring, aliphatic chain, and piperidine ring. A number of PPR derivatives with modified piperine have been reported to show pharmacologic properties, including inhibition of NorA efflux pump (Kumar et al., 2008), anticancer, and antibacterial activity (Rama Subba Rao et al., 2012; Umadevi et al., 2013).
P450 (CYP) enzymes are the major enzymatic system responsible for elimination of many pharmaceutical agents (Lee et al., 2012). Inhibition of CYP enzymes may result in serious consequences, particularly for medicinal agents with narrow therapeutic window (VandenBrink and Isoherranen, 2010; Parkinson et al., 2011). The inhibition of CYP enzymes can be roughly divided into two types: direct inhibition by the parent compounds and metabolism-dependent inhibition (MDI) by metabolites (Lee et al., 2012). MDI includes reversible, quasi-irreversible, and irreversible inhibition (Lin and Lu, 1998). The latter two are classified as mechanism-based inhibition (MBI). Methylenedioxyphenyl (MDP) compounds are an important class of chemicals widely occurring in insecticides, oils, and spices (Murray, 2000). As early as 1970, MDP compounds were found to enhance the toxicity of insecticides, which was reportedly achieved by inhibiting the mixed-function oxidase system of microsomes (Casida, 1970). Many compounds containing such structures are microsomal substrates as well as inhibitors and have been documented to cause CYP inactivation. Alkaloids canadine, bulbocanine, and carnitine were reported to be mechanism-based and quasi-irreversible inactivators of CYP2C19 (Salminen et al., 2011). Myristicin is a mechanism-based inactivator of CYP1A2, and the reactive metabolites involved in the enzyme inactivation are quinone tautomers (Yang et al., 2015). Five kinds of MDP lignans found in piper cubeba were documented to be irreversible inhibitors of CYP3A4 reportedly resulting from their metabolic activation (Usia et al., 2005). Two reactive metabolites/intermediates derived from MDP-containing compounds are suggested to be responsible for the host enzyme inactivation, including catechol metabolites and carbene intermediates. The carbene intermediates can coordinate with the heme of CYP enzymes. The formation of carbene-heme-iron-porphyrin complexes results in quasi-irreversible inhibition of CYPs (Taxak et al., 2013). The catechols can be further oxidized to reactive ortho-quinones which sequentially alkylate apoprotein of CYPs (Hutzler et al., 2006, 2008).
Intake of foods may induce the expression or inhibit the activity of CYP enzymes, which may interfere with the pharmacokinetic properties of various drugs. For instance, grapefruit juice ingestion can specifically inhibit CYP3A4-mediated drug metabolism (Ohnishi et al., 2000). Furanocoumarin epoxybergamottin, a component squeezed from the peel of grapefruits into the juice during industrial manufacturing, was found to demonstrate inhibitory activity on CYP3A (Wangensteen et al., 2003). CYP3A is the major P450 subfamily extensively distributed in hepatocytes and intestine epithelial cells (Rendic and Di Carlo, 1997; Murray, 2000).
In this study, we studied the inhibitory effect of PPR on human hepatic CYP enzymes and characterized the reactive metabolites possibly involved in enzyme inactivation. The significance of the present study is to understand the biochemical mechanism of the interaction of CYP enzymes with PPR, which is important to the justification of potential food-drug interactions resulting from PPR to ensure the safety and effectiveness of medications.
Materials and Methods
Chemicals.
Piperine (PPR) with purity >98% was acquired from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). Superoxide dismutase (SOD) was purchased from Shanghai Jianglai Biologic Technology Co., Ltd. (Shanghai, China). Propranolol, bupropion·HCl, dextromethorphan, tolbutamide, coumarin, testosterone, 4-nitrophenol, phenacetin, α-naphthoflavone, pilocarpine, methoxsalen, sulfaphenazole, ticlopidine, quinidine, disulfiram, ketoconazole, NADPH, glutathione (GSH), N-acetylcysteine (NAC), and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) were obtained from Sigma-Aldrich (St. Louis, MO). Mixed human liver microsomes prepared from adult male donors and recombinant human CYP enzymes were provided by BD Gentest (Woburn, MA). All organic solvents and reagents were analytical or high-performance liquid chromatography grade and purchased from Fisher Scientific (Springfield, NJ). DMSO-d6 was purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA).
Chemical Synthesis of DM-PPR.
PPR (4.0 mg) and 8.0 mg of anhydrous AlCl3 were placed in a 25 ml round-bottom flask and mixed with 3.0 ml of toluene. The mixture was then refluxed at 110°C for 12 hours, followed by incorporation of additional anhydrous AlCl3 (2.0 mg) and another 12 hours of refluxing. The resulting mixture was mixed with 0.5 ml of water after being cooled to room temperature, and shaken well. The resulting mixture was concentrated under vacuum. The residue was submitted to semipreparative liquid chromatography for purification, and purified DM-PPR (Scheme 1) was characterized by mass spectrometry (MS) on Bruker hybrid quadrupole-time-of-flight mass spectrometer and NMR on a BRUKER-ARX-600 spectrometer (Bruker Corporation, Carteret, NJ).

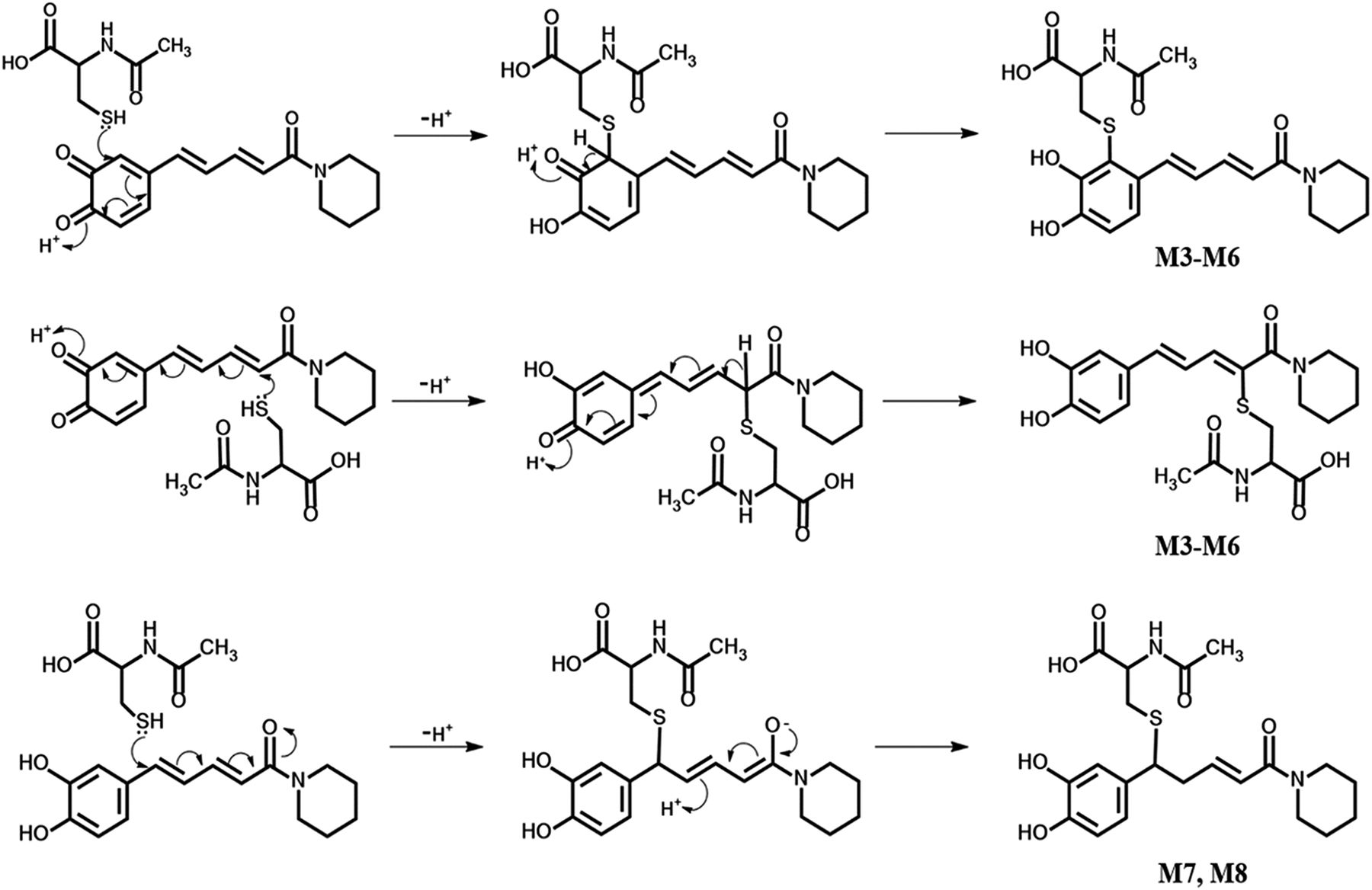
Proposed pathways for the generation of reactive intermediate(s) and NAC conjugates derived from PPR.
Chemical Synthesis of NAC Conjugates.
NAC conjugates M1 and M2 were synthesized as follows: PPR (200 μM) was directly mixed with NAC (20 mM) in phosphate-buffered saline (PBS) (100 mM, 2 ml), followed by stirring at 37°C for 60 minutes and centrifugation at 19,000g. The supernatants were analyzed by liquid chromatography coupled to tandem MS (LC-MS/MS). For synthesis of NAC conjugates M3–M8, DM-PPR (1.0 mg, 3.7 μmol) was dissolved in 2.0 ml of CH2Cl2. After mixed with DDQ (2.0 mg, 8.8 μmol), the solution was stirred at 4°C for 4 hours. The organic solvent was removed by evaporation, and the resulting crude products were redissolved in 2.0 ml of water. The mixture was further reacted with NAC (4.0 mg, 25 μmol) with stirring at room temperature for 12 hours. The final mixture was centrifuged, and the supernatants (5.0 μl) were subjected to LC-MS/MS for analysis.
Screening of Time-Dependent CYP Inhibition by PPR.
The time-course inhibitory effects of PRR on CYPs were determined by monitoring individual CYP activities of human liver microsomes at various time points after being exposed to PPR. The primary incubation mixtures contained PPR (0 or 80 μM), MgCl2 (3.2 mM), and human liver microsomes (0.5 mg protein/ml) in 200 μl PBS (100 mM, pH = 7.4). After preincubation at 37°C for 3 minutes, NADPH (1.0 mM) was added to the primary incubations to initiate the reactions. After the primary mixtures were incubated for 0, 5, 10, and 20 minutes, 40 μl aliquots were transferred to the secondary reaction mixtures (120 μl) composed of individual probe substrates (100 μM bupropion for CYP2B6, 50 μM phenacetin for CYP1A2, 5.0 μM dextromethorphan for CYP2D6, 120 μM tolbutamide for CYP2C9, 200 μM 4-nitrophenol for CYP2E1, 200 μM coumarin for CYP2A6, or 200 μM testosterone for CYP3A), MgCl2 (3.2 mM), and NADPH (1.0 mM), and further incubation for 15 minutes, respectively. Ice-cold acetonitrile (120 μl) containing internal standard (IS) propranolol (100 ng/ml) was mixed with the incubation solution to quench the reactions. The resulting supernatants after vortex-mixing and centrifuging were analyzed by LC-MS/MS. The loss of enzyme activities was determined by monitoring the generation of the metabolites resulting from the probe substrates applied in the secondary incubations.
Determination of Time-, Concentration-, and NADPH-Dependent Inhibition of CYP3A.
The primary incubation mixtures (200 μl) were composed of PPR at concentrations of 0, 5.0, 10, 20, 40, 60, or 80 μM, MgCl2 (3.2 mM), and human liver microsomes (0.5 mg protein/ml) in PBS (100 mM, pH 7.4). After 3 minute preincubation, the reaction was initiated by the addition of NADPH (1.0 mM). At time points of 0, 5, 10, and 20 minutes, 40 μl of the primary incubations was withdrawn for CYP3A activity assessment in the secondary incubations that contained testosterone (200 μM) and NADPH (1.0 mM) in PBS (100 mM, pH 7.4). After a 15 minutes incubation, the reactions were quenched using ice-cold acetonitrile (120 μl) containing propranolol (100 ng/ml) as IS. The supernatants were analyzed by LC-MS/MS after being centrifuged at 19,000g for 10 minutes. To determine NADPH-dependence of the observed CYP3A enzyme inhibition, PBS, which replaced NADPH with the same volume was included in microsomal incubations with PPR (80 μM).
Determination of Time- and NADPH-Dependent Inhibition of CYP3A by DM-PPR.
The inhibitory effect of DM-PPR on CYP3A was evaluated, and similar protocol as above was executed except for the replacement of PPR with DM-PPR (80 μM). Control incubations lacked DM-PPR or NADPH.
Determination of Effect of Ketoconazole on PPR-Mediated Inactivation of CYP3A.
Ketoconazole is a competitive inhibitor of CYP3A. The primary incubation system contained PPR (80 μM), MgCl2 (3.2 mM), human liver microsomes (0.5 mg protein/ml), and ketoconazole at concentrations of 0, 0.01, 0.1, and 1.0 μM. The procedures of subsequent reaction initiation/termination and sample preparation were the same as described above.
Determination of Effects of SOD/Catalase and GSH on PPR-Mediated Inactivation of CYP3A.
Nucleophile glutathione (GSH, 2.0 mM) was included in the primary mixture together with PPR (80 μM), MgCl2 (3.2 mM), human liver microsomes (0.5 mg protein/ml), and NADPH (1.0 mM). At 0 and 20 minutes, the resulting incubation mixtures (40 μl for each) were transferred to the secondary incubations containing probe substrate testosterone for the evaluation of the remaining enzyme activities. In a separate study, similar incubations were performed except incorporation of superoxide dismutase (SOD) and catalase (800 unit/ml for each) to replace GSH. The control groups lacked either GSH or SOD/catalase.
Determination of Partition Ratio.
To determine the partition ratio, PPR at final concentrations of 0, 2.0, 5.0, 10, 20, 40, 60, 80, and 100 μM and human recombinant CYP3A4 (50 nM) were mixed in the primary reactions. NADPH (1.0 mM) was added to initiate the reactions. Aliquots (40 μl) were taken at time points of 0 and 20 minutes to the secondary incubation mixtures containing testosterone as a probe substrate for measuring the remaining CYP3A4 activities.
Determination of Reversibility of PPR-Mediated CYP3A Inhibition through Dialysis.
A mixture of PPR (0 or 80 μM), human liver microsomes (0.5 mg protein/ml), and NADPH (1.0 mM) was incubated at 37°C for 0 and 20 minutes. The resulting mixture was transferred into Slide-A-Lyzer cassettes (molecular mass cutoff: 3500 Da; Pierce, Rockford, IL) and dialyzed in 900 ml of PBS (100 mM, pH 7.4) at 4°C three times for 2 hours each time. In parallel, nondialyzed samples were allowed to stand at 4°C for 6 hours. Aliquots (40 μl) of all samples were submitted to the secondary incubations (120 μl) composed of testosterone as a probe substrate after being brought to room temperature, respectively. CYP3A enzyme activities of the resulting mixtures were determined as stated below.
Determination of Reversibility of PPR-Mediated CYP3A Inhibition by Potassium Ferricyanide.
Whether carbene intermediate was involved in PPR-mediated CYP3A inhibition was determined by following the reported methods (Watanabe et al., 2007; Hong et al., 2015). Briefly, the primary incubations containing PPR (0 or 80 μM), human liver microsomes (0.5 mg/ml), and NADPH were executed at 37°C. After 0 or 20 minutes incubation, the resulting solutions (40 μl) were mixed with K3Fe(CN)6 (0 or 2.0 mM) dissolved in10 μl of PBS (100 mM, pH 7.4). After 10 minutes incubation, the mixtures were subjected to secondary incubation for assessment of the remaining CYP3A activities as below.
P450 Enzyme Assays.
The amounts of the products formed from individual probe substrates were measured to evaluate the corresponding CYP enzyme activities by an AB Sciex 5500 triple-quadrupole mass spectrometer (Applied Biosystems, Foster City, CA) equipped with an Agilent 1260 Series Rapid Resolution HPLC (Agilent Technologies, Santa Clara, CA). A Promosil C18 column (150 × 4.6 mm, 5 μm; Agela Technologies, Inc., Tianjin, China) was employed for analyte separation. The solvent system was composed of mobile phase A (water containing 0.1% formic acid) and phase B (acetonitrile with 0.1% formic acid). The flow rate was maintained at 0.8 ml/min. The gradient elution was set as follows: 1) 0–2.0 minutes, 90% A; 2.0–2.5 minutes, 90%–70% A; 2.5–14.0 minutes, 70%–30% A; 14.0–14.1 minutes, 30%–10% A; 14.1–15.0 minutes, 10%–90% A; 15.0–17.0 minutes, 90% A for the products of CYP2B6, 2D6, and 1A2; 2) 0–2.0 minutes, 90% A; 2.0–5.0 minutes, 90%–30% A; 5.0–10.0 minutes, 30%–5% A; 10.0–12.0 minutes, 5% A; 12.0–13.0 minutes, 5%–90% A; 13.0–15.0 minutes, 90% A for the products of CYPs2C9, 2E1, and 2A6; and 3) 0–3.0 minutes, 70% A; 3.0–8.0 minutes, 70%–66% A; 8.0–10.0 minutes, 66%–10% A; 10.0–11.0 minutes, 10% A; 11.0–11.1 minutes, 10%–70% A; 11.1–13.0 minutes, 70% A for the product of CYP3A. Multiple reaction monitoring (MRM) scan mode was applied to quantitate the products. Ion pairs of MRM acquired included the following: 152.1→110.0 for acetaminophen (CYP1A2), 256.0→238.0 for hydroxybupropion (CYP2B6), 287.2→171.2 for 4-hydroxytolbutamide (CYP2C9), 258.2→157.1 for dextrorphan (CYP2D6), 305.3→269.4 for 6β-hydroxytestosterone (CYP3A), and 260.3→116.3 for propranolol (IS) in positive mode, 161.0→105.1 for 7-hydroxycoumarin (CYP2A6), and 153.9→122.8 for 4-nitrocatechol (CYP2E1) in negative mode.
Metabolite Identification.
PPR (100 μM), human liver microsomes (1.0 mg protein/ml), MgCl2 (3.2 mM), NAC (10 mM), and NADPH (1.0 mM) were mixed in 100 μl PBS (100 mM, pH 7.4) and incubated at 37°C for 30 minutes. NADPH was not included in negative control. The resulting reactions were processed as above, followed by LC-MS/MS analysis. The mobile-phase system included water (A) and acetonitrile (B), both containing 0.1% formic acid. And the column was eluted with a gradient program as follows: 0–2.0 minutes, 80% A; 2.0–9.0 minutes, 80%–55% A; 9.0–13.0 minutes, 55% A; 13.0–14.0 minutes, 55%–10% A; 14.0–15.0 minutes, 10% A; 15.0–16.0 minutes, 10%–80% A; 16.0–18.0 minutes, 80% A. An AB SCIEX Instruments 4000 hybrid triple quadrupole-liner ion trap (Q-Trap) mass spectrometer was applied to characterize NAC conjugates. MS/MS analyses were executed through an MRM-EPI (enhanced product ion)scanning in positive-ion mode (449.0→320.0 for M1 and M2, 435.0→221.0 for M3–M6, and 437.0→181.1 for M7 and M8).
Metabolizing Enzyme Inhibition Studies.
To determine which P450 enzymes preferentially catalyze the metabolism of PPR, PPR was incubated with human liver microsomes in the presence of individual selective CYP inhibitors. Similar microsomal incubations as described above were conducted with the minor modification that CYP inhibitors were included in microsomal mixtures besides the components listed above. The inhibitors applied, along with their final concentrations, were α-naphthoflavone (10 μM for 1A2), methoxsalen (20 μM for 2A6), sulfaphenazole (20 μM for 2C9), ticlopidine (100 μM for 2B6 and 2C19), quinidine (10 μM for 2D6), disulfiram (10 μM for 2E1), and ketoconazole (10 μM for 3A) (referring to the FDA Guidance for Industry on Drug Interaction Studies). The control group did not include the inhibitors. Sample preparation and product quantification were performed as above. P450 activities were calculated as the percentage of the control activities, each incubation was carried out three times in three separate days.
Recombinant Human P450 Incubations.
Similar incubations were conducted as above microsomal study, and PPR was incubated with individual recombinant human P450 enzymes (100 nM in each incubation), including CYPs2A6, 2C9, 1A2, 2E1, 3A4, 2C19, 3A5, 2B6, and 2D6, in place of human liver microsomes. The resulting NAC adducts (M3–M8) were analyzed and quantified by LC-MS/MS. The formation of NAC adducts (%) was calculated by determination of the ratio of peak area of metabolites in individual recombinant CYP enzymes to the most abundance of the metabolites generated.
Statistical Analysis.
All of the data reported represent the mean ± S.D. Statistical evaluation was performed using unpaired t test, and *P < 0.05, **P < 0.01, and ***P < 0.001 were accepted to be statistically significant.
Results
Overview of PPR-Mediated Time-Dependent Inhibition of P450 Enzymes.
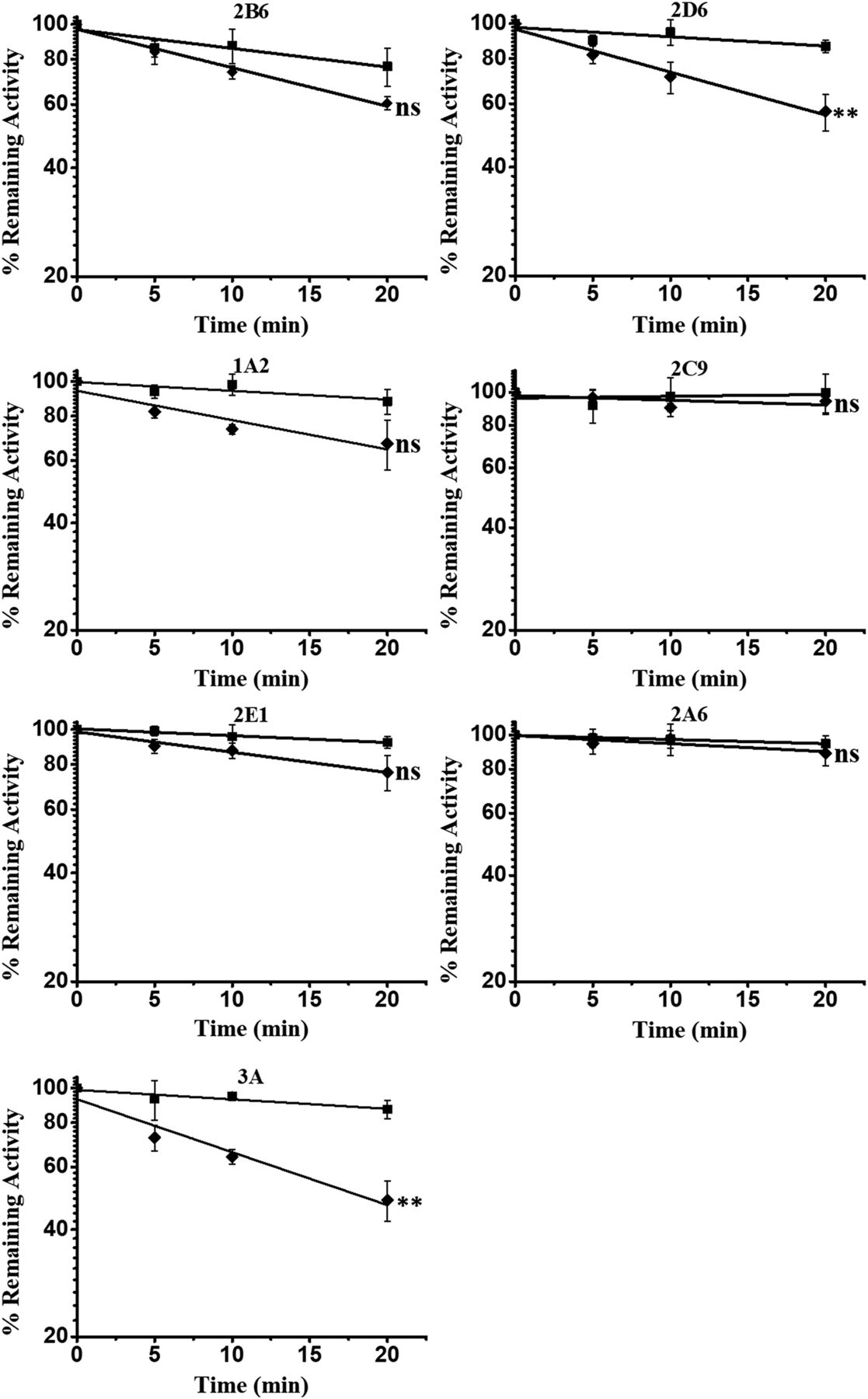
In order to define the inhibitory effect of PPR (80 μM) on seven common CYP enzymes, we first performed a quick screening study. The remaining activities of the seven CYP enzymes were measured after incubation at various time points. As illustrated in Fig. 1, loss of CYPs3A and 2D6 activities was observed after 20 minutes incubation. The remaining enzyme activities of CYP3A and CYP2D6 were about 50% and 57%, respectively.

Quick screening of time-dependent inhibition of cytochrome P450 enzymes by PPR. PPR at concentrations of 0 (■) or 80 (◆) μΜ, human liver microsomes (0.5 mg protein/ml), and NADPH (1.0 mM) were incubated at 37°C for 0, 5, 10, and 20 minutes. The residual enzymatic activities at 0 minute were normalized to 100%. Data represent the mean ± S.D. (n = 3). Comparisons were made using an unpaired t test. * indicates the significance of the remaining enzyme activity of experimental groups with respect to that of control at the same time point (20 minutes) of incubations. Levels are considered significant at *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
Time-, Concentration-, and NADPH-Dependent Inhibition of CYP3A by PPR.
As shown in Fig. 2A, CYP3A activity decreased with incubation time after exposure to PPR at concentrations of 5.0, 10, 20, 40, 60, or 80 μM. Specifically, exposure to PPR at 80 μM in 20 minutes at 37°C resulted in a loss of approximately 50% CYP3A activity (Fig. 2A). No such enzyme inactivation was observed in the absence of either PPR or NADPH (Fig. 2B) under the same incubation condition.

(A) Time- and concentration-dependent inhibition of CYP3A by PPR. PPR at concentrations of 0 (■), 5 (●), 10 (▲), 20 (▼), 40 (◆), 60 (★), and 80 (○) μM was incubated with human liver microsomes (0.5 mg protein/ml) fortified with NADPH (1.0 mM) at 37°C for 0, 5, 10, and 20 minutes. (B) NADPH-dependent inactivation of CYP3A4 by PPR. Human liver microsomes (0.5 mg/ml) were incubated with vehicle (■) or PPR (80 μM) in the absence (▼) or presence (★) of NADPH. (C) Wilson’s plot. kobs values were calculated from the slope of the regression lines shown in (A). Data represent the mean ± S.D. (n = 3). * indicates the significance of the remaining enzyme activity of experimental groups with respect to that of control at the same time point (20 minutes) of incubations. $ indicates the significance of the remaining enzyme activity of experimental groups at various incubation times, compared with that at 0 minute under the same incubation condition. # indicates the significance of the remaining enzyme activity in the presence of NADPH, compared with that in the absence of NADPH under the same incubation condition. Levels are considered significant at *P < 0.05; **/$$/##P < 0.01; ***P < 0.001; ns, not significant.
A double-reciprocal plot (Wilson’s plot) of the observed rates of inactivation (kobs) and PPR concentrations above were applied to calculate kinetic constants KI and kinact (Fig. 2C). KI was obtained from the negative reciprocal of the x-axis intercept, and kinact was determined by the value of the 2.3/y-axis intercept (Alvarez-Diez and Zheng, 2004). Therefore, KI and kinact were calculated to be 30.7 μM and 0.041 minutes−1, respectively.
Protection Effect of Competitive Inhibitor on PPR-Mediated CYP3A Inactivation.
Ketoconazole was coincubated with PPR in human liver microsomes. The protective effect of ketoconazole increased with the increase in concentrations of ketoconazole applied (Fig. 3). The residual enzymatic activity of CYP3A in the absence of ketoconazole was 47.9% ± 5.2% at 20 minutes, while the remaining enzyme activities were 59.3% ± 0.7%, 71.6% ± 1.9%, and 81.0% ± 1.3% in the presence of ketoconazole at concentrations of 0.01, 0.1, and 1.0 μM, respectively.

Effect of competitive inhibitor on inactivation of CYP3A by PPR. Human liver microsomes (0.5 mg protein/ml) were incubated with vehicle (■) or PPR (80 μM) supplemented with ketoconazole at concentrations of 0 (★), 0.01 (◆), 0.1 (▲), or 1.0 (●) μM. Data represent the mean ± S.D. (n = 3). * indicates the significance of the remaining enzyme activity of experimental groups with respect to that of control at the same time point (20 minutes) of incubations. Levels are considered significant at *P < 0.05; **P < 0.01; ***P < 0.001.
Effects of GSH and SOD/Catalase on Enzyme Inactivation.
The effects of GSH and SOD/catalase on enzyme inactivation were determined by examining PPR-mediated CYP3A inactivation in the absence or presence of GSH as well as SOD/catalase. After incubation for 20 minutes, the remaining activities of CYP3A were 55.1% ± 1.1% with GSH (2.0 mM) and 52.5% ± 0.7% without GSH. SOD and catalase, scavengers of reactive oxygen species (ROS), displayed little protective effects on the inactivation of CYP3A, and the remaining enzyme activities were 53.0% ± 3.5% (with SOD/catalase) and 52.5% ± 0.7% (without SOD/catalase) after 20 minute incubations.
Partition Ratio for Inactivation of CYP3A4 by PPR.
Partition ratio (P value) is a measure of the efficiency of a mechanism-based inactivator. The P value was calculated by a plot of the remaining enzyme activity versus the PPR/CYP3A4 molar ratio. As shown in Fig. 4, two linear regression lines were seen, with PPR/CYP3A4 as the abscissa and the remaining enzyme activity as the ordinate. The P value was 273 calculated from P + 1 of 274 estimated based on the value of the intersection of two linear regression lines.

Partition ratio of CYP3A4 inactivation by PPR. CYP3A4 (50 nM) was incubated with PPR at designed concentrations (0, 2, 5, 10, 20, 40, 60, 80, or 100 μM). The extrapolated P + 1 value was determined from the point of intersection to the abscissa. Data represent the mean ± S.D. (n = 3).
Reversibility of Inactivation.
The dialysis experiment was designed and executed to determine the reversibility of the observed CYP3A enzyme inhibition after exposure to PPR. The changes in CYP3A activity of microsomes exposed to PPR and those treated with vehicle before and after dialysis were examined. Apparently, the observed loss of CYP3A activity was reversed by 6 hours dialysis, and the enzyme activity was restored from 28.2% ± 3.2% (predialysis) to 71.9% ± 5.2% (post-dialysis), about 43.7% of CYP3A activity was recovered.
Effect of Ferricyanide on PPR-Mediated CYP3A Inhibition.
The contribution of carbene intermediate to enzyme inactivation was investigated by selective oxidation of carbene-iron complex with potassium ferricyanide. Compared with the vehicle group, 39.7% of CYP3A activity was recovered when PPR-pretreated (80 μM) microsomes were incubated with K3Fe(CN)6 (2.0 mM) (Table 1).
Effects of K3Fe(CN)6 on the inactivation of CYP3A
Data represent the mean ± S.D. (n = 3). The percentage of CYP3A activity was determined by calculation of the ratio of enzyme activity of each sample after 0 or 20 minute incubation with PPR vs. that of the corresponding control sample without PPR treatment. % Reversed stands for the contribution of KFC to the recovered activity of CYP3A.
Formation of Quinone Metabolite from PPR.
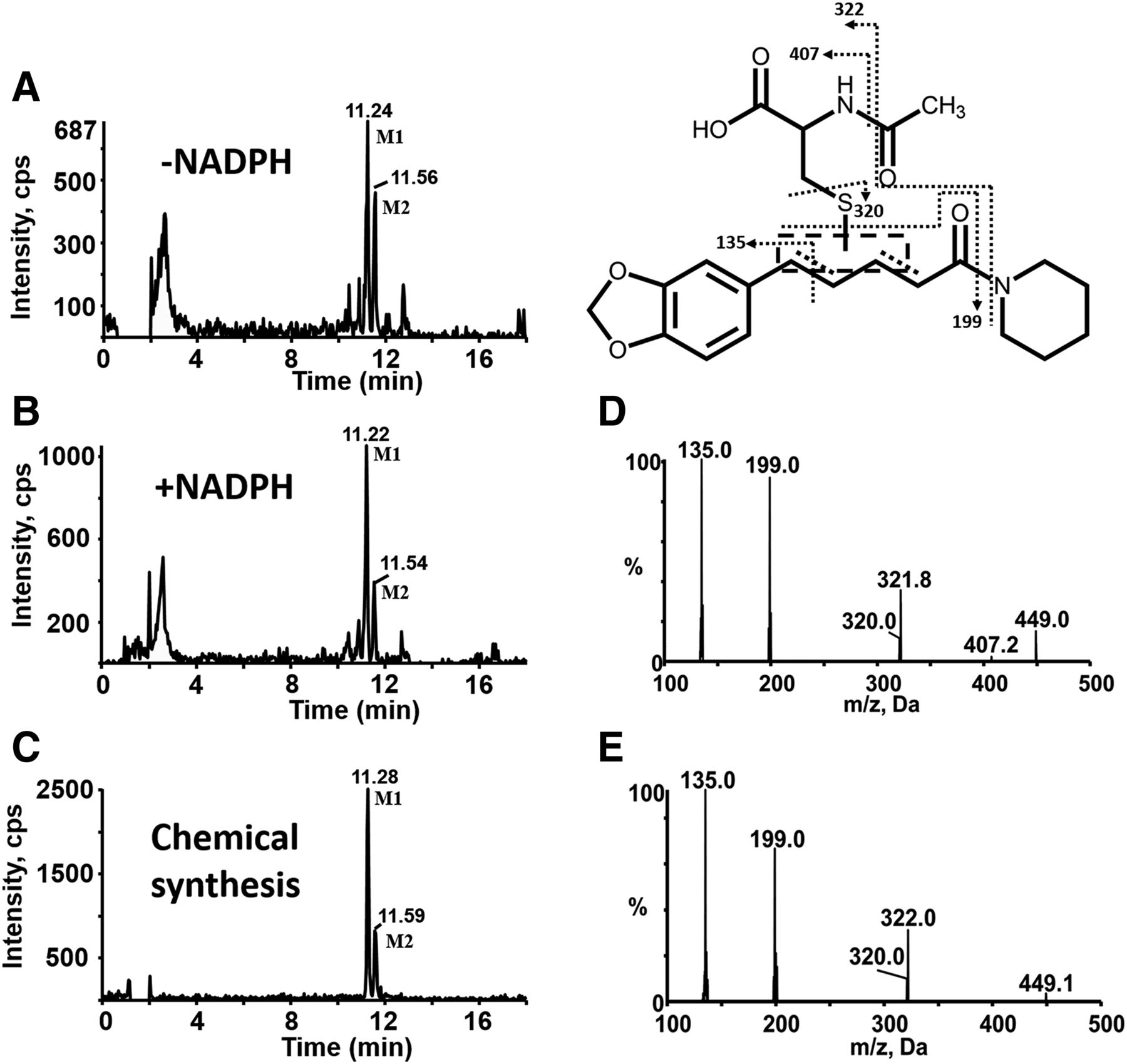
Characterization of reactive metabolites of PPR was carried out in human liver microsomes supplemented with NAC as a trapping agent. A total of eight NAC conjugates (M1–M8) were found in the resulting microsomal mixture. Metabolites M1 and M2 with [M + H]+ ion at m/z 449 eluted at 11.22 and 11.54 minutes, respectively (Fig. 7B). And their mass spectra showed representative fragment ions related to the NAC moiety (Fig. 7D). The product ion at m/z 320 was obtained from the neutral cleavage of the C−S bond of the NAC moiety (−129 Da). The fragment ion at m/z 407 originated from the loss of the acetyl portion (−42 Da) from m/z 449. Product ion at m/z 322 arose from the loss of the piperidine ring (−85 Da) from m/z 407. The fragment ion at m/z 135 resulted from the cleavage of PPR portion. In addition, M1 and M2 were detected in microsomal incubations both in the absence and presence of NADPH (Fig. 7, A and B).Fig 5

Chemical structure and 1H NMR chemical shifts of DM-PPR.
Another two types of NAC adducts with molecular ions m/z 435 (M3-M6) and 437 (M7, M8) were detected (Fig. 8B; Fig. 9B, respectively; Schemes 1 and 2). Four peaks assigned as M3 (retention time = 10.49 minutes), M4 (retention time = 10.99 minutes), M5 (retention time = 11.39 minutes), and M6 (retention time = 11.72 minutes) (Fig. 8B) showed their [M + H]+ at m/z 435, in agreement with the molecular weight of DM-PPR–derived NAC conjugates. The generation of M3–M6 was NADPH-dependent (Fig. 8, A and B). MRM-EPI of ion transition at m/z 435/221 was scanned to acquire the MS/MS spectra of the four NAC conjugates. Identification of M3–M6 was based on the major fragments at m/z 306, 263, and 221 (Fig. 8D). Fragment ion m/z 306 came from the characteristic neutral loss of 129 Da (the NAC moiety) from m/z 435, and fragment ion m/z 221 was derived from the loss of piperidine ring (−85 Da) from m/z 306. The product ion at m/z 263 resulted from the loss of the acetyl, carboxyl, and piperidine ring of the parent ion.Fig 5

Proposed mechanisms for the formation of NAC conjugates M3–M8.
In addition to M3–M6, another type of DM-PPR-derived NAC conjugates, namely, M7 and M8, with retention time at 10.99 and 11.37 minutes (Fig. 9B) were detected in the above microsomal incubations. Incubations in the absence of NADPH did not produce the two conjugates (Fig. 9A). The molecular ion of conjugates M7–M8 was m/z 437, which is 2 Da (2 H) higher than that of M3–M6 (m/z 435). MRM-EPI scanning (ion transition m/z 437/181) revealed representative fragment ions (Fig. 9D), including m/z 308 (loss of 129 Da) associated with the indicative neutral loss of the NAC portion, m/z 223 derived from the further loss of piperidine ring (−85 Da) from m/z 308, and m/z 181 generated from the loss of −CO (28 Da) and −CH2 (14 Da) of m/z 223.
Chemical synthesis was carried out to verify the structure of the metabolites detected in microsomal incubations. M1–M8 were all observed in the synthetic mixture by LC-MS/MS. The resulting products showed the same chromatographic (Fig. 7C, Fig. 8C, Fig. 9C) and mass spectrometric (Fig. 7E; Fig. 8E; Fig. 9E) identities as those generated in microsomal incubations. Unfortunately, poor reaction yield and heavy impurity with similar retention time mad us fail to obtain sufficient amounts of the individual products for NMR characterization.
Time- and NADPH-Dependent Inhibition of CYP3A by DM-PPR.
To probe further probe the contribution of carbene intermediate to the inhibition of CYP3A, DM-PPR was synthesized and tested. High-resolution mass spectrometry analysis of synthetic DM-PPR revealed its [M + H]+ of m/z 274.1436, consistent with the corresponding theoretical mass within 5 ppm, compared with the predicted formula (Supplemental Table 1). We succeeded in obtaining 1H NMR (DMSO-d6, 600 MHz) spectrum of synthetic DM-PPR (Supplemental Fig. 1), and the proton assignment is listed in Fig. 5. The signals responsible for the methylene moiety observed in the 1H NMR spectrum of the parent compound disappeared in the spectrum of the product.
DM-PPR at concentrations of 0 and 80 μM, was incubated with human liver microsomes in the absence or presence of NADPH and displayed time-dependent inhibition of CYP3A. NADPH was required for enzyme inhibition (Fig. 6A). Exposure to DM-PPR at 80 μM for 20 minutes at 37°C resulted in about a 31.8% ± 2.5% loss of CYP3A activity (Fig. 6), while exposure to the parent compound at the same molar concentration resulted in a 50.3% ± 2.7% loss (Fig. 6B) of enzyme activity under the same condition.

(A) Time- and NADPH-dependent inhibition of CYP3A by DM-PPR (80 μM). Human liver microsomes (0.5 mg protein/ml) were incubated with vehicle (■), or DM-PPR (80 μM) in the absence (▲) or presence (◆) of NADPH at 37°C for 0, 5, 10, and 20 minutes. (B) Human liver microsomes (0.5 mg protein/ml) were incubated with vehicle (■), DM-PPR (80 μM) (◆), or PPR (80 μM) (★) in the presence of NADPH. The residual enzymatic activities at 0 minute were normalized to 100%. Data represent the mean ± S.D. (n = 3). * indicates the significance of the remaining enzyme activity of experimental groups with respect to that of control at the same time point (20 minute) of incubations. (A) # indicates the significance of the remaining activity in the presence of NADPH, compared with that in the absence of NADPH under the same incubation condition; (B) # indicates the significance of the remaining activity of the incubation containing DM-PPR (80 μM), compared with that of PPR (80 μM) in the presence of NADPH under the same conditions. ##P < 0.01; and ***P < 0.001 are considered significantly different; ns, not significant.

MS characterization of metabolites M1 and M2. Extracted ion (m/z 449.0/320.0 for M1 and M2) chromatograms obtained from LC-MS/MS analysis of incubations containing PPR, human liver microsomes, and NAC in the absence (A) or presence of NADPH (B). (C) Extracted ion chromatograms obtained from LC-MS/MS analysis of synthetic M1 and M2. (D) MS/MS spectra of M1 and M2 generated in microsomal incubations. (E) MS/MS spectra of synthetic M1 and M2.
P450 Enzymes Responsible for PPR Bioactivation.
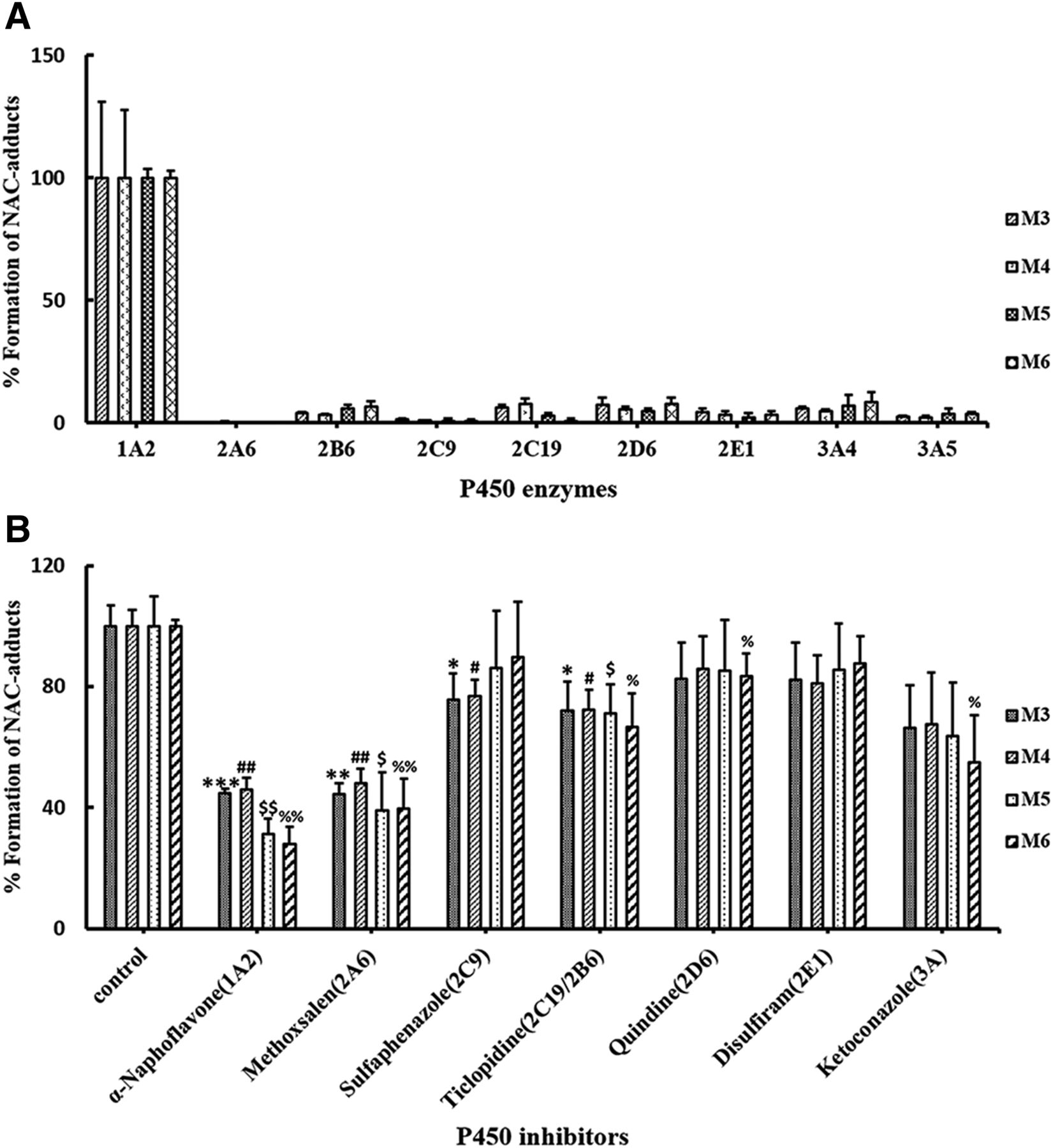
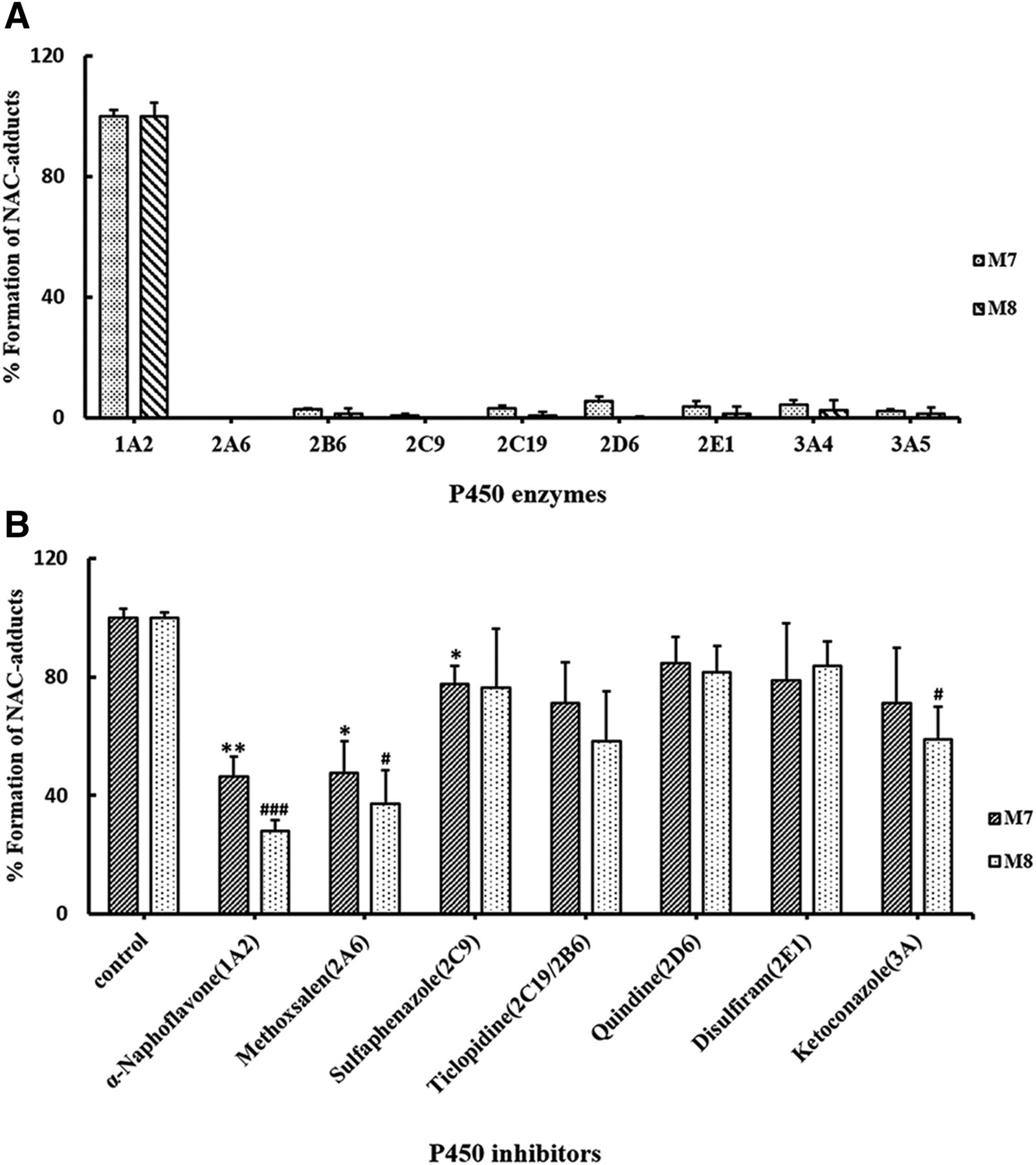
To identify which P450 enzymes are mainly responsible for the bioactivation of PPR, nine human recombinant CYPs were individually incubated with PPR, NAC, and NADPH, followed by assessment of the formation of M3–M6 and M7, M8. P450 1A2 was the major enzyme participating in the formation of M3–M6 and M7, M8 (Fig. 10A; Fig. 11A).
Additionally, PPR was incubated with human liver microsomes fortified with NAC in the absence or presence of individual P450 enzyme inhibitors. As shown in Figs. 10B and 11B, α-naphthoflavone, inhibitor of CYP 1A2, elicited inhibitory effects on the production of M3–M6 and M7, M8. Specifically, the production of M3–M6 and M7, M8 remained at levels of 55.2% ± 1.6%, 54.0% ± 3.9%, 68.6% ± 4.9%, 72.0% ± 5.8%, 53.5% ± 6.5%, and 72.0% ± 3.6%, respectively.
Discussion
The rapid screening experiment demonstrated that PPR caused significant loss of CYP3A and CYP2D6 activity after 20 minutes of microsomal incubation (Fig. 1). Apparently, PPR inhibited CYP3A the most, which encouraged us to investigate the interaction of PPR with CYP3A as a model enzyme.
PPR was found to cause time-, concentration-, and NADPH-dependent inhibition of CYP3A (Fig. 2, A and B). The observed requirement of NADPH for PPR-mediated CYP3A inhibition indicates that metabolic activation was involved in the enzyme inhibition. Competitive inhibitor coincubation experiments showed that ketoconazole reduced the enzyme inactivation of CYP3A by PPR (Fig. 3). This result suggests that the bioactivation of PPR occurred at the active site of CYP3A, and the competitive binding of ketoconazole in the active site of the host enzyme protected the enzyme from inactivation (Moruno-Davila et al., 2001).
In consideration of reactive intermediates that release from the active site of the host enzyme and of ROS produced during metabolism also can contribute to enzyme inactivation, the effects of GSH and scavengers of ROS on enzyme inactivation were determined. GSH revealed minor protection against enzyme inactivation by PPR. This indicates that the reactive intermediates produced via P450-mediated metabolism resided the pocket of the host enzyme. SOD and catalase, scavengers of ROS, displayed little protective effect on the inactivation of CYP3A, That implies that ROS, if produced, was not involved in the enzyme inactivation.
Partition ratio (P value) is a measure to evaluate the effectiveness of a mechanism-based inactivator. The less the value is, the more effective the inactivator. The reported P values of mechanism-based inactivators ranged from almost zero to several thousands (Kent et al., 2001). The observed P value of PPR was approximately 273 (Fig. 4), which can be classified as a moderately efficient inactivator of CYP3A4.
Dialysis experiments were conducted to define the interaction of the metabolite of PPR with host enzyme CYP3A. Apparently, dialysis restored the enzyme activity of CYP3A, indicating that the binding mode of the enzyme to the metabolite was noncovalent.
MDP compounds can be metabolized to carbene intermediates mediated by P450 enzymes, and the resulting carbene may coordinate with the iron of the heme, resulting in inhibition of the host enzymes (Buening and Franklin, 1976; Muakkassah et al., 1982). Potassium ferricyanide can reportedly reverse the carbene-mediated inhibition by selective oxidation and dissociation of carbene-heme-iron-porphyrin complexes (Buening and Franklin, 1976). The addition of K3Fe(CN)6 showed a protective effect on the inhibition of CYP3A by PPR in the incubation (Table 1), indicating that the formation of the carbene-iron complex was possibly involved in CYP3A inactivation.
Alternatively, the resulting carbene may be converted to the corresponding formate ester, The formate ester may be hydrolyzed to catechol and further oxidized to the corresponding ortho-quinone (Scheme 1) (Murray, 2000). As expected, we succeeded in detection the NAC conjugate derived from the putative quinone intermediate in human liver microsomal incubations (Fig. 8; Scheme 2). In addition, NAC conjugates M3–M8 were detected in human liver microsomal incubations with synthetic DM-PPR (Supplemental Fig. 3, A and B). This suggests P450-mediated formation of the quinone intermediate proposed in Scheme 1. To determine the contribution of the quinone intermediate in PPR-mediated CYP3A inactivation, we evaluated the inhibitory effect of DM-PPR and found that DM-PPR showed weak enzyme inactivation relative to that caused by PPR (Fig. 6). Whether the enzyme inhibition by DM-PPR is reversible was determined by dialysis. As expected, exhaustive dialysis failed to restore the DM-PPR inhibited CYP3A activity (the residual CYP3A activities were 46.7% ± 7.4% without dialysis and 52.8% ± 3.9% with dialysis). Taken together, the observed CYP3A inactivation resulted not only from carbene-mediated iron coordination but also from quinone-derived protein modification.

MS characterization of DM-PPR–derived NAC conjugates M3-M6. Extracted ion (m/z 435.0/221.0 for M3-M6) chromatograms obtained from LC-MS/MS analysis of incubations containing PPR, human liver microsomes, and NAC in the absence (A) or presence of NADPH (B). (C) Extracted ion chromatograms obtained from LC-MS/MS analysis of synthetic M3–M6. (D) MS/MS spectra of M3–M6 generated in microsomal incubations. (E) MS/MS spectra of synthetic M3–M6.

MS characterization of DM-PPR–derived NAC conjugates M7 and M8. Extracted ion (m/z 437.0/181.0 for M7 and M8) chromatograms obtained from LC-MS/MS analysis of incubations containing PPR, human liver microsomes, and NAC in the absence (A) or presence of NADPH (B). (C) Extracted ion chromatograms obtained from LC-MS/MS analysis of synthetic M7 and M8. (D) MS/MS spectra of M7 and M8 generated in microsomal incubations. (E) MS/MS spectra of synthetic M7 and M8.

P450 enzymes responsible for the generation of M3–M6 in vitro. (A) Rates of M3–M6 formation in incubations containing PPR, individual recombinant P450 enzymes, NAC, and NADPH. (B) Effects of selective P450 inhibitors on the generation of M3–M6. PPR was incubated with human liver microsomes, NAC, and NADPH, in the absence (control) or presence of individual P450 enzyme inhibitors. Data represent the mean ± S.D. (n = 3). *, #, $, and % indicate the significance of the amount of M3–M6 produced in experimental groups compared with that of the control, respectively. Comparisons were made using unpaired t test. */#/$/%P < 0.05; **/##/$$/%%P < 0.01; ***/###/$$$/%%%P < 0.001 are considered significantly different.

P450 enzymes responsible for the generation of M7 and M8 in vitro. (A) Rates of M7 and M8 formation in incubations containing PPR, individual recombinant P450 enzymes, NAC, and NADPH. (B) Effects of selective P450 inhibitors on the generation of M7 and M8. PPR was incubated with human liver microsomes, NAC, and NADPH in the absence (control) or presence of individual P450 enzyme inhibitors. Data represent the mean ± S.D. (n = 3). * and # indicate the significance of the amount of M7 and M8 produced in experimental groups, compared with that of the control, respectively. Comparisons were made using unpaired t test. */#P < 0.05; **P < 0.01; ###P < 0.001 are considered significantly different.
Structurally, PPR is a Michael acceptor (unsaturated ketone). As expected, two NAC conjugates (M1 and M2) were detected in human liver microsomal incubations, possibly resulting from 1,4- to 1,6-addition (Scheme 1). Additionally, cysteine conjugates were observed in PPR-exposed recombinant CYP3A4 after proteolytic digestion (Supplemental Fig. 2). Thus, we cannot exclude the possibility that the observed enzyme inactivation arose from nonspecific covalent modification of CYP3A via the Michael addition. This prompted us to determine the potential involvement of the nonspecific modification in the enzyme inactivation. As a result, we failed to see enzyme inactivation in the absence of NADPH. This suggests that the nonspecific protein addition, if it took place, did not necessarily cause enzyme inactivation.
Both recombinant and microsomal inhibition studies verified that CYP1A2 was the primary enzyme participating in the bioactivation of PPR measured by monitoring the formation of NAC adducts derived from the quinone intermediate (M3–M8) (Figs. 10 and 11). Methoxsalen was reportedly an inhibitor of both CYPs 2A6 and 1A2 (Ono et al., 1996). That is probably why the results obtained from the CYP2A6 recombinant incubations and microsomal inhibition incubations did not quite match each other. Interestingly, CYP1A2 was found to be the major enzyme which mediated the bioactivation of PPR, but PPR caused significant inactivation of CYP3A and slight inactivation of CYP2D6. Reactive metabolites generated in the active site of CYP3A selectively inactivated the host enzymes, which made their activity not surviving long enough to produce many metabolites. Additionally, reactive metabolites generated in the active site of enzymes did not necessarily result in inactivating the host enzymes, and it also depends on the availability of nucleophilic amino acid residues at their active sites and the suitable distance between the resulting reactive intermediate and the targeting nucleophilic amino acid residues of the host enzymes. For some reason, the reactive metabolite generated in situ was unable to modify the amino acid residue(s) in the active site of CYP1A2, sequentially escaped from the host enzymes, and trapped by NAC. Whether PPR is a mechanism-based inactivator of CYP2D6 needs further investigation.
In summary, PPR quasi-irreversibly inhibited CYP3A, and biotransformation was required for the enzyme inactivation. Both the carbene and ortho-quinone reactive intermediates are involved in the inactivation of CYP3A.
Authorship Contributions
Participated in research design: Cui, Peng, Zheng.
Conducted experiments: Cui, Wang, Tian, Zhang.
Performed data analysis: Cui.
Wrote or contributed to the writing of the manuscript: Cui, Zheng.
Footnotes
- Received August 7, 2019.
- Accepted November 5, 2019.
↵1 Y.P. and J.Z. contributed equally to this work. The two corresponding units contributed equally to this work.
This work was supported in part by the National Natural Science Foundation of China [Grants 81830104, 81430086, 81773813, and U1812403].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- DDQ
- 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DM-PPR
- demethylene piperine
- EPI
- enhanced product ion
- GSH
- glutathione
- IS
- internal standard
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- MBI
- mechanism-based inactivator
- MDP
- methylenedioxyphenyl
- MRM
- multiple reaction monitoring
- NAC
- N-acetylcysteine
- P450
- cytochrome P450
- PBS
- phosphate-buffered saline
- PPR
- piperine
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}