


Abstract
Organic anion transporting polypeptides (OATPs), expressed in human liver (OATP1B1, OATP1B3, and OATP2B1) and intestine (OATP2B1), govern the pharmacokinetics (PK) of drugs (e.g., statins) and endogenous substrates (e.g., coproporphyrin I [CPI]). Their expression is known to be modulated (e.g., disease, age, and environmental factors), and they also present as the loci of clinically relevant polymorphisms and drug interactions involving inhibition. In comparison, relatively few clinical reports describe the induction of OATPs, although the effect of inducers (e.g., rifampicin [RIF], carbamazepine [CBZ]) on OATP biomarker plasma levels and statin PK has been reported. Of note, available human tissue (e.g., biopsy) protein and messenger RNA expression profiling data indicate that OATPs in gut and liver are not induced by prototypical inducers such as RIF when compared with cytochrome P450 3A4 (CYP3A4), P-glycoprotein (Pgp), multidrug resistance–associated protein 2 (MRP2), and breast cancer resistance protein (BCRP). Such results are consistent with in vitro human hepatocyte data. Therefore, the observed impact of RIF, and possibly CBZ, on statin PK (>20% decrease in the area under the plasma concentration vs. time curve) cannot be ascribed to OATP induction with certainty. In fact, most statins and CPI have been shown to present variously as substrates of RIF-inducible proteins such as CYP3A4, Pgp, MRP2, and BCRP. Interpretation of multidose RIF data is further complicated by its autoinduction, which likely leads to decreased inhibition of OATP. In the absence of more conclusive OATP induction data, caution is needed when modeling drug-drug interactions involving multidose inducers such as RIF.
SIGNIFICANCE STATEMENT Presently, there is limited direct clinical evidence supporting the notion that human liver and gut organic anion transporting polypeptides (OATPs) are inducible by agents like rifampicin (RIF). Such data need to be reconciled and will pose challenges for attempting to incorporate OATP induction into physiologically based pharmacokinetics models. Although disparate sets of tissue biopsy (atorvastatin and carbamazepine) and in vitro hepatocyte (phenobarbital, chenodeoxycholate, and amprenavir) data present OATP messenger RNA induction (≥2-fold) by agents beyond RIF, the clinical relevance of such data needs to be determined.
Introduction
Of the various solute carriers (SLCs) known to be expressed in the human liver, two organic anion transporting polypeptides (OATPs), OATP1B1 and OATP1B3, are well characterized. A third OATP (OATP2B1) is ubiquitously expressed including the intestine and liver (Oscarson et al., 2006, 2007; Brueck et al., 2019). To a greater or lesser degree, each of the three is known to be involved in the pharmacokinetics (PK) of various drugs (e.g., statins, sartans, gliptins), endogenous compounds (e.g., coproporphyrin isomers coproporphyrin I [CPI] and CPIII, bilirubin glucuronide, and amidated bile acid glucuronides and sulfates), and agents supporting liver function testing and imaging (de Graaf et al., 2011; Yoshikado et al., 2016; Rodrigues et al., 2018; Mori et al., 2019). Any combination of the three OATPs can also serve as the target of important drug-drug interactions (DDIs) involving inhibition (Poirier et al., 2007; Yoshida et al., 2012; Jamei et al., 2014; Vaidyanathan et al., 2016). Beyond DDIs, the expression and function of individual OATPs in human tissues can be modulated by genetic polymorphisms (e.g., loss-of-function alleles), proinflammatory mediators, viral infections, cholestatic drugs, and metabolic diseases such as nonalcoholic steatohepatitis (Le Vee et al., 2009; Gong and Kim, 2013; Clarke et al., 2014; Billington et al., 2018; Vildhede et al., 2019).
One aspect of clinical research that has received relatively little attention, when compared with drug-metabolizing enzymes (e.g., cytochrome P450 3A4 [CYP3A4]) and various ATP-binding cassette (ABC) transporters (e.g., P-glycoprotein [Pgp], multidrug resistance–associated protein 2 [MRP2, ABCC2], and breast cancer resistance protein [BCRP]), is the induction of OATPs by nuclear receptor agonists such as rifampicin (RIF) and carbamazepine (CBZ). Although regulation of OATP expression and function has been the subject of reviews (Svoboda et al., 2011; Staudinger et al., 2013; Alam et al., 2018), nuclear receptor–mediated OATP induction has been somewhat underrepresented, with some researchers studying the effect of OATP expression and genotype on the exposure of inducers and possible impact on the induction of CYP3A4. This is because inducers like RIF and hyperforin are known to be OATP substrates, which may govern their intracellular concentration and interaction with nuclear receptors such as the pregnane X receptor (PXR, NR1I2) (Tirona et al., 2003; Niemi et al., 2006a; Schäfer et al., 2019).
Perspective
It is accepted that RIF induces CYP3A4 and ABC transporters via the PXR, whereas CBZ (a relatively weak PXR agonist vs. RIF) manifests a somewhat different induction signature via other nuclear receptors such as the constitutive androstane receptor (CAR, NR1I3) (Faucette et al., 2007; Kim et al., 2010). In fact, RIF has been studied extensively as an inducer in vitro and in animals (humanized rodents and nonhuman primate), and clinical assessment has involved the use of CYP3A biomarkers (e.g., 6β-hydroxycortisol–to-cortisol urine ratio, plasma 4β-hydroxycholesterol) as well as CYP3A (e.g., midazolam) and Pgp (e.g., digoxin, dabigatran etexilate) probe drugs (Greiner et al., 1999; Rae et al., 2001; Peng et al., 2011; Li et al., 2014; Henderson et al., 2019; Tahara et al., 2019; Yamazaki et al., 2019). Because various statins are accepted clinical probes for the study of SLCO1B1 genotype-phenotype associations and inhibitory OATP DDIs, it is not surprising that there are numerous reports describing the impact of multidose RIF and CBZ on their PK (Kyrklund et al., 2003; Ucar et al., 2004; Backman et al., 2005; Chung et al., 2006; Lutz et al., 2018a,b). More recently, this has extended to the OATP biomarker CPI (Kunze et al., 2018).
At face value, one could interpret such changes in statin PK (e.g., >20% decrease in the area under the statin plasma concentration vs. time curve [AUC]) as induction of OATP in the gut and liver and then consider such information when attempting to model multidose DDIs for new molecular entities in development (Table 1). Likewise, because plasma CPI is increasingly considered a viable OATP1B1/3 biomarker (Rodrigues et al., 2018), it is only natural to assume that decreases in its plasma concentration following RIF multiple dosing could also be reflective of OATP induction (Kunze et al., 2018). However, the predose CPI levels in plasma are not impacted by multiple doses of RIF, which indicates that OATP induction is unlikely. The decrease in maximal CPI plasma concentration following RIF dosing is more likely reflective of reduced RIF exposure, following autoinduction, that results in loss of OATP inhibition (Kunze et al., 2018).
Reported impact of RIF and CBZ on the PK of putative OATP statin probes and biomarker CPI
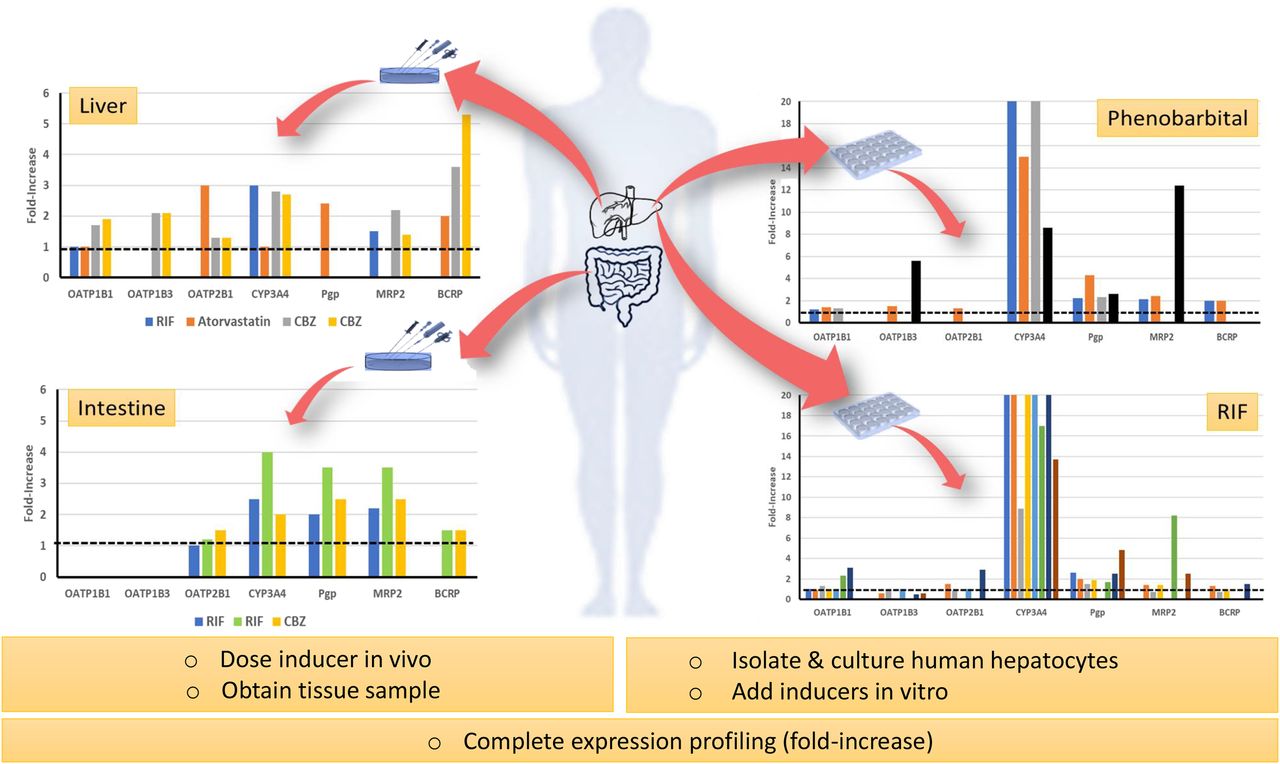
From the standpoint of expression profiling, in vitro and human tissue biopsy data argue against clinically meaningful induction of OATPs following a probe inducer such as RIF (Fig. 1). In fact, could one make the case that documented multidose DDIs are largely driven by induction of CYP3A4 and ABC transporters in the gut and liver? At the same time, although RIF is widely studied and serves as the pharmaceutical industry’s gold standard potent inducer, it is subject to autoinduction involving unidentified mechanisms (Acocella, 1978). This will further complicate the modeling of its PK and DDIs. Admittedly, there are caveats when interpreting relatively weak induction of OATPs in vitro or analyzing data from human tissues. For example, one assumes that biopsy data are reflective of the whole organ and that maximal induction (Emax) ratios (e.g., CYP3A4-to-OATP1B1 or Pgp-to-OATP1B1 Emax ratio) obtained in vitro faithfully reflect the situation in vivo.

Summary of available published in vitro (human hepatocytes) and ex vivo (gut and liver tissue) expression profiling data for human OATP forms, CYP3A4, and various ABC transporters following an inducer. Summary of data presented in Table 3 (tissue profiling) and Table 4 (cultured human hepatocytes and human liver tissue slices). Colors represent different publications (in vitro data) or inducers (tissue expression data) referenced in summary tables.
Although we focused on human data, we do acknowledge that some investigators have explored the induction of various OATP forms expressed in animals (Rausch-Derra et al., 2001; Cheng et al., 2005; Niu et al., 2019). Recent data published by Niu et al. (2019) are particularly interesting because they deployed the cynomolgus monkey as a model to investigate the induction of OATP by RIF. The authors concluded that RIF, a known cynomolgus monkey PXR agonist and CYP3A4 inducer, does not induce cynomolgus monkey liver OATP1B1 and OATP1B3.
In the following, we discuss evidence supporting and refuting the induction of human gut and liver OATPs; clinical DDI data are described, in the context of the extended clearance classification system (ECCS), in addition to in vitro induction data and the results of human tissue expression profiling following inducer administration.
Regulation of OATP Expression and Function
Both the transcriptional and posttranslational regulation of OATP expression and function have already been reviewed extensively by others (Svoboda et al., 2011; Staudinger et al., 2013; Murray and Zhou, 2017; Alam et al., 2018). With respect to gene expression, certain OATP gene promoter regions have been characterized, transcription factor and nuclear receptor (e.g., PXR, CAR, vitamin D receptor, farnesoid X receptor [FXR, NR1H4], and liver X receptor alpha [LXRα, NR1H3]) binding motifs identified, and their impact on expression in cell-based assays has been studied (Jung et al., 2001, 2002; Wood et al., 2005; Meyer zu Schwabedissen and Kim, 2009; Meyer zu Schwabedissen et al., 2010; Eloranta et al., 2012).
Like many SLCs, human OATP1B1 and OATP1B3 are known to be glycosylated, undergo ubiquitination, and to be subject to kinase-dependent phosphorylation. All serve to modulate their function, intracellular cycling, turnover, and trafficking to and from the plasma membrane (Murray and Zhou, 2017; Xu and You, 2017; Alam et al., 2018). This implies that OATP mRNA expression does not necessarily correlate with protein expression and function. Recently, Zhang and Hagenbuch (2019) have proposed that, once resident on plasma membranes, individual SLCs can form heterodimers and homodimers and form higher order structures that could be further regulated. Hypothesis testing and in vitro–in vivo extrapolations are further complicated by additional human OATPs (e.g., intestinal OATP3A1, OATP4A1, and OATP4C1) that are poorly characterized in terms of their regulation, response to inducers, and role in DDI (Oswald, 2019). Even for well-characterized OATPs, such as OATP1B1 and OATP1B3, the role of microRNAs and epigenetics (e.g., histone methylation and acetylation) in governing their expression and function must be considered (Kacevska et al., 2011, 2012; Rieger et al., 2013; Krattinger et al., 2016). Therefore, a new chemical entity could modulate basal OATP expression in a given tissue by any number of mechanisms.
Such complexity necessitates the development and careful validation of more sophisticated in vitro test systems and human-relevant animal models to support new chemical entity screening and in vitro–to–in vivo extrapolations. As described in the following, of critical importance is the consideration of OATP induction (by perpetrator) in the context of CYP3A and ABC transporter induction, as well as the role of OATP in governing the overall clearance of the victim drug and its susceptibility to such induction.
Interpreting and Modeling of Clinical DDI Data and the Challenge of Using RIF as a Model Inducer
A case can be made that the PK of statins is governed not solely by OATP but also by combinations of various ABC transporters expressed in the gut and liver; nearly all statins more or less present as Pgp, BCRP, and MRP2 substrates in vitro, and some present as CYP3A4 or cytochrome P450 2C (CYP2C) substrates (Jacobsen et al., 1999; Prueksaritanont et al., 1999; Yeo and Yeo, 2001; Chen et al., 2005; Huang et al., 2006; Knauer et al., 2010; Gupta et al., 2016; Shin et al., 2017; Afrouzian et al., 2018). This is particularly true for atorvastatin and rosuvastatin, whose PK is associated with both SLCO1B1 and BCRP (ABCG2) genotype (Niemi, 2010). Of the statins, simvastatin is known to be extensively metabolized by CYP3A4 (Table 2), which is highly inducible by RIF (vs. OATP) in both the gut and liver (Table 3). Even so-called “selective” OATP probes like pravastatin show limited oral absorption because of ABC transporter-mediated efflux. For example, pravastatin is a known MRP2 substrate, which is inducible in gut and liver by RIF (Tables 2 and 3). In agreement, Shen et al. (2018) have reported that itraconazole has a minimal impact on CPI plasma levels indicative of weak OATP1B1 and OATP1B3 inhibition in vivo. However, itraconazole has been shown to increase the AUC of oral pravastatin by as much as 72% when dosed prior to the statin. Could this implicate ABC transporters? Notably, itraconazole also increases the AUC of digoxin, a widely accepted Pgp substrate (Supplemental Table 1).
In vitro data presenting various OATP probes as CYP3A and ABC transporter substrates
Summary of some literature reports describing OATP expression profiling of human tissues after administration of an inducer
It can be argued that the single dose PK of an OATP probe, such as pravastatin, is not associated with ABC transporter genotype (e.g., ABCG2) (Niemi, 2010). However, following an RIF induction regimen (600 mg every day, ≥7 days), there is ample evidence indicating that the expression of ABC transporters is increased and likely leads to increased biliary and intestinal secretory clearance. Such a hypothesis, involving the induction of MRP2, has been proposed by Kyrklund et al. (2003) to explain why multidosing of RIF decreases the plasma AUC (∼30%) of oral pravastatin in their study. Relatedly, Niemi et al. (2006b) reported a 68% decrease in pravastatin AUC in subjects carrying the ABCC2 c.1446C>G allele, which reflects a nonsynonymous nucleotide polymorphism (threonine at position 482) on exon 10 that leads to an ∼2.0-fold increase in liver MRP2 mRNA expression. The authors did not assess the impact on ABCC2 genotype on intestinal MRP2 expression. As discussed in the following, such an increase in liver MRP2 expression (in the absence of OATP1B1 induction) is comparable to that reported by Marschall et al. (2005) following RIF multidosing. Overall, this implies that statins can be used as single dose OATP phenotyping tools or as probes to assess OATP inhibition in the absence of induction (i.e., following a single perpetrator dose). However, statins’ use in clinical studies to investigate OATP induction per se should be questioned. The same could apply to CPI, also an MRP2 substrate, as an OATP biomarker.
RIF as Inducer and Autoinducer
Although RIF is well studied as an inducer, in many ways it remains an enigmatic drug. For example, it is an inducer of numerous drug-metabolizing enzymes beyond CYP3A4 and presents as an OATP and ABC transporter substrate in vitro (Rae et al., 2001; Tirona et al., 2003; Spears et al., 2005; Poirier et al., 2014a,b), and its inhibition of gut CYP3A4 and Pgp is a consideration when modeling and designing DDI studies involving induction (Reitman et al., 2011; Baneyx et al., 2014; Supplemental Table 2). At the same time, although its absorption-distribution-metabolism-excretion profile in human subjects has been documented, it is only recently that the enzymes involved in its metabolism have been identified. RIF is now known to be largely metabolized by microsomal arylacetamide deacetylase (AADAC), expressed in the gut and liver, to the 25-desacetyl RIF metabolite (Nakajima et al., 2011; Kobayashi et al., 2012). This major metabolite is recovered (with parent RIF) in urine and bile (Acocella, 1978). A second relatively minor metabolite (3-formyl RIF) is thought to be formed nonenzymatically and may itself undergo AADAC-dependent metabolism. Currently, there is no direct evidence that RIF is a CYP3A substrate, and AADAC is not inducible (at least in human hepatocytes in vitro) (Nishimura et al., 2002; N. Johnson, Pfizer, Inc., unpublished results). So why does RIF (e.g., 600 mg every day) manifest autoinduction after multiple dosing (Acocella, 1978)?
RIF does present as an OATP1B1 (initially reported as OATP-C) and OATP1B3 (initially reported as OATP8) substrate, not as an OATP2B1 (initially reported as OATP-B) substrate (previously unpublished Pfizer data in Supplemental Table 3;Vavricka et al., 2002; Tirona et al., 2003), but we are making a case here that none of these SLCs is significantly induced in vivo. On the other hand, as described above, RIF does present as a Pgp and MRP2 substrate in vitro, and it does appear to significantly induce both ABC transporters in vivo (Table 3). Therefore, could a case be made that RIF autoinduction is the result of its induction of gut and liver Pgp and MRP2 expression, which leads to increased gut secretion and biliary clearance? Because both RIF metabolites (25-desacetyl RIF and 3-formyl RIF) are also MRP2 and Pgp substrates (Supplemental Table 3), this implies that their clearance would also be increased after multidosing of RIF. The increased recovery of parent RIF and 25-desacetyl RIF in the bile of subjects dosed 600 mg for 7 days (vs. day 1) is consistent with this hypothesis (Acocella, 1978). We noted that publications ascribed the observed autoinduction to “increased metabolism” of RIF in both the gut and liver (Acocella, 1978; Loos et al., 1987; Chirehwa et al., 2015). In hindsight, are the results of such studies reporting increased biliary and gut secretion of RIF and its metabolites via Pgp and/or MRP2? In agreement, Smythe et al. (2012) have posed a hypothesis that RIF autoinduction could be explained by PXR-mediated induction of Pgp in the gut and liver.
OATP Induction in the Context of PK Modeling
Given the large number of induction studies with RIF, it is not surprising that the wealth of clinical data has spawned numerous publications describing various RIF model files to support PK and physiologically based PK (PBPK) modeling (Reitman et al., 2011; Yamashita et al., 2013; Baneyx et al., 2014). Although numerous groups have modeled CYP3A induction, more recent PBPK models have been expanded to include induction of gut Pgp, liver CYP2C8, CYP2C9, and OATP (Asaumi et al., 2018, 2019; Hanke et al., 2018; Yamazaki et al., 2019).
In particular, the recent report by Asaumi et al. (2019) caught our attention. In this instance, the authors expanded their original RIF PBPK model (Asaumi et al., 2018) to incorporate OATP1B (OATP1B1 and OATP1B3) induction (Emax = 2.2–2.3) akin to CYP2C8 (Emax = 2.6) and assigned a concentration of inducer at half of Emax (EC50) for OATP1B based on CYP3A4. It is noteworthy that additional authors claim to have similarly developed an extended PBPK model for RIF and have considered its Pgp (Emax = 2.5), AADAC (Emax = 0.99), CYP3A4 (Emax = 9.0), CYP2C8 (Emax = 3.2), OATP1B1 (Emax = 0.38), and OATP1B3 (Emax = 0.38) induction signature based on available biopsy data and in vitro data (Hanke et al., 2018; Türk et al., 2019). A major limitation is that there is no consensus regarding the best RIF Emax values to use as input (or for reconciling the observed DDIs) for OATP1B1 and OATP1B3 in PBPK models. Unlike OATP1B1 and OATP1B3, there is closer agreement across groups regarding the model RIF Emax input values for Pgp, CYP3A4, and CYP2C8 (Reitman et al., 2011; Yamashita et al., 2013; Baneyx et al., 2014; Asaumi et al., 2018, 2019; Hanke et al., 2018; Yamazaki et al., 2019). On the contrary, Varma et al. (2013b, 2014) were able to describe the effect of time staggering of the RIF dose on the AUC of OATP1B, CYP3A, and CYP2C8 substrates (repaglinide and glyburide) by simply adopting CYP3A induction (Emax and EC50) and an OATP inhibition constant obtained in vitro.
DDIs Involving Induction by RIF in the Context of ECCS
Concepts involving liver clearance have evolved to include both transporter and enzymatic activity. Depending on the rates of the individual transport (active and passive transport) and metabolic processes, an “extended clearance” term may be applied to identify the rate-determining step in the overall hepatic clearance of a drug. It is generally acceptable to assume “rapid-equilibrium” conditions between blood and liver compartments for highly permeable compounds that are not substrates of an uptake transporter. In such a scenario, metabolism is typically the rate-determining step in hepatic clearance. However, when uptake via transporters such as OATP1B1 and OATP1B3 is involved, hepatic clearance can be “uptake-determined” or influenced by “transporter-enzyme interplay” (Varma et al., 2015; Kimoto et al., 2018). Therefore, OATP induction can differentially impact victim drugs manifesting rapid-equilibrium hepatic clearance, OATP uptake-determined hepatic clearance, or OATP-enzyme interplay. However, factors such as CYP and ABC transporter induction in the gut and liver need to be considered also.
To identify any previously unrecognized case examples in support of OATP induction in vivo, we evaluated clinical DDIs per ECCS class following chronic treatment with RIF (Fig. 2). An extensive data set was developed with about 200 substrate drugs for which AUC ratio values (i.e., AUC without and with RIF treatment) were available. According to the ECCS framework—substrate drugs categorized based on their ionization, molecular weight, and permeability (Varma et al., 2015)—metabolism is suggested to be the primary driver of systemic clearance for high permeability basic and neutral compounds (class 2). In the case of high permeability, low molecular weight (≤400 Da) acidic or zwitterionic compounds (class 1A), organic anion transporter 2–enzyme interplay contributes to hepatic clearance (Kimoto et al., 2018). On the other hand, OATP1B-mediated hepatic uptake is often the primary systemic clearance mechanism for high permeability, high molecular weight (>400 Da) acidic or zwitterionic compounds (class 1B).

Clinical DDIs per ECCS class involving a prototypic inducer such as RIF. AUC ratio of substrate drug dosed intravenously (A) and orally (B) following chronic oral RIF treatment. Open data points represent the AUC ratio of individual substrate drug; and box and whiskers depict median and upper and lower quartile with error bars representing range. Closed data points are mean values. Shaded area represents no induction, whereas horizontal green and red lines denote boundaries for no interaction (AUC ratio 0.8–1.25), as well as a weak (AUC ratio >0.5), moderate (AUC ratio 0.5–0.2), and strong (AUC ratio <0.2) induction effect. Data set was built after exhaustive and careful mining of published literature using the Metabolism and Transport Drug Interaction Database (DIDB; www.druginteractioninfo.org). ECCS class assignment was similar to that previously reported (Varma et al., 2015). For permeability classification of substrate drugs into ECCS, apparent permeability was measured across Madine-Darby canine kidney cells selected for low endogenous transporter expression. Drug ionization state was assigned based on calculated pKa values using MoKa (version 2.5.4; Molecular Discovery Ltd).
ECCS Class 1B.
Consistent with the ECCS-based assignment of the clearance mechanisms, RIF-based DDIs are predominant in class 2 with about 40% of substrate drugs showing strong interactions (AUC ratio <0.2). On the contrary, strong interactions are limited in other classes with a few exceptions, including atorvastatin and repaglinide (class 1B). Other class 1B drugs such as bosentan, glyburide, and macitentan present weak-to-moderate interactions following oral RIF treatment. Given these four drugs are predominantly metabolized by CYP3A, induction of intestinal metabolism likely contributes to the noted AUC changes. This can be substantiated in the case of atorvastatin because its oral exposure has been shown to be unaltered by intravenous itraconazole but increased ∼3-fold following oral itraconazole (Kantola et al., 1998; Maeda et al., 2011). Similar conclusions can be inferred based on the mechanistic modeling and simulations for repaglinide and glyburide (Varma et al., 2013a, 2014).
ECCS Class 3A and 4.
Low permeability, low molecular weight (≤400 Da) acidic or zwitterionic compounds (class 3A) and low permeability basic and neutral compounds (class 4) are primarily subjected to renal clearance. However, given the low passive permeability, these drugs are subjected to intestinal efflux via ABC transporters such as Pgp, BCRP, and MRP2, which may limit their oral absorption. Albeit with a small sample size, only pefloxacin showed some change in its systemic exposure following RIF treatment. Pgp-mediated efflux is thought to limit its oral absorption (fraction absorbed ∼60%); thus, induction of intestinal Pgp may explain the AUC ratio of ∼0.57 following RIF treatment (Griffiths et al., 1994). Similarly, weak-to-moderate RIF induction effects were also noted for class 4 drugs. Metabolically stable Pgp substrates with limited oral absorption such as aliskiren, celiprolol, dabigatran etexilate, ranitidine, and talinolol represent this class; consequently, induction of intestinal efflux further limits their oral exposure.
ECCS Class 3B.
Finally, class 3B represents low permeability high molecular weight (>400 Da) acidic or zwitterionic compounds, which are primarily cleared by OATP1B-mediated hepatic uptake and/or cleared unchanged in urine. In this instance, moderate changes in AUC are noted for OATP1B substrate drugs, including fexofenadine, rosuvastatin, and simeprevir. However, clinical evidence implies that the PK of these poorly absorbed drugs is affected by efflux inhibitors and ABCG2 genotype. Although drugs such as fexofenadine and rosuvastatin involve OATP2B1-mediated intestinal absorption, the net effect of RIF treatment points toward induction of intestinal efflux transporters (Table 3).
Profiling of Human Tissue after Dosing with Inducer
RIF
It was possible to find five references describing CYP3A4, ABC transporter, and OATP human tissue expression profiling following treatment with an inducer (Fig. 1; Table 3). Four of the reports described the biopsy of study subjects before and after inducer. The work of Marschall et al. (2005) is particularly important because it is one of the few examples of liver biopsy following an inducer such as RIF. In this instance, healthy patients with gallstones (scheduled for cholestectomy) were randomized to RIF (600 mg/d for 1 week), ursodeoxycholic acid (1 g/d for 3 weeks), or no medication before surgery. A liver biopsy specimen was taken to study the expression of transporters and drug-metabolizing enzymes. As expected, CYP3A4 mRNA was induced (3-fold) by RIF, which corresponded to a 247% increase in plasma biomarker levels (4β-hydroxycholesterol) in the same (biopsied) subjects. In contrast, ursodeoxycholic acid did not induce CYP3A4 mRNA and elicited less impact on plasma 4β-hydroxycholesterol levels (38% increase). Of note, RIF induced MRP2 mRNA and protein expression (∼2-fold) but did not induce OATP1B1 mRNA expression. Unfortunately, the study did not evaluate the expression of Pgp, BCRP, and additional OATPs in the samples. Lack of induction of OATP1B1 by ursodeoxycholic acid is somewhat anticipated because it presents as a weak FXR agonist (vs. chenodeoxycholate) in human hepatocytes (Liu et al., 2014).
According to Oscarson et al. (2007) and Brueck et al. (2019), RIF also elicits weak induction (≤1.2-fold) of OATP mRNA in the human gut (e.g., OATP2B1), which is somewhat surprising given the anticipated high concentrations of the inducer during first pass (Baneyx et al., 2014; Asaumi et al., 2018). Presumably, RIF interacts with PXR in the gut because the expression of CYP3A4, Pgp, and MRP2 in duodenal biopsies is induced (2.0–4.0-fold).
CBZ
To our knowledge, tissue expression profiling following oral CBZ is reported twice (Oscarson et al., 2006; Brueck et al., 2019); CBZ is known to interact with CAR and is a weak PXR agonist (vs. RIF). Like RIF, tissue expression data point to CBZ as a relatively weak inducer of intestinal OATP2B1 when compared with CYP3A4, Pgp, MRP2, and BCRP (Brueck et al., 2019; Table 3). However, data provided by Oscarson et al. (2006) for two epileptic subjects on CBZ are compelling. Although they used a small sample set, the authors were able to show that hepatic OATP1B1 and OATP1B3 mRNA expression was induced ∼2-fold in both subjects (vs. the livers of n = 7 control subjects); the induction was comparable to that observed for CYP3A4 and MRP2 mRNA but lower than for BCRP mRNA (3.6-, 5.3-fold).
Atorvastatin
To date, perhaps one of the most intriguing reports describing expression profiling of liver biopsy samples is by Björkhem-Bergman et al. (2013). The authors reported that OATP2B1 mRNA was statistically significantly induced (3-fold; P < 0.05) in liver biopsies of subjects receiving atorvastatin (80 mg for 4 weeks). In the same study, an ∼2-fold induction of liver Pgp and BCRP was noted in the absence of induction of OATP1B1 and CYP3A4 mRNA. Fluvastatin (20 mg/d for 4 weeks) was dosed in the same study but elicited a relatively minimal effect on hepatic gene expression profiles. Unfortunately, the above-described biopsy data are discordant with the results of in vitro studies supporting that atorvastatin is a PXR agonist and induces CYP3A4 mRNA (∼6-fold) in human primary hepatocytes (Hoffart et al., 2012). The exact mechanism of how atorvastatin elicits induction of hepatic OATP2B1 in vivo, without induction of OATP1B1 or CYP3A4, is not known and needs confirmation. However, it is known that the OATP2B1 gene promoter region is distinct from that of OATP1B1 and OATP1B3, which explains its broader tissue expression profile and possibly its differentiated induction signature (Maeda et al., 2006).
In Vitro Data
We also turned our attention to various publications describing studies with cryopreserved human primary hepatocytes in culture (coculture or sandwich) and precision-cut human liver tissue slices rather than hepatic (e.g., HepG2, HepaRG, and Huh7) or intestinal (e.g., Caco-2, LS180, T84 and LS174T) cell lines. Unfortunately, reports of RIF incubation with primary human enterocytes, as well as precision-cut human intestinal slices, have largely focused on CYP3A4 and ABC transporters (van de Kerkhof et al., 2008; Li et al., 2018).
Plated Human Primary Hepatocytes
Examples of reports describing studies with human primary hepatocytes in culture are shown in Table 4 and focus as much as possible on papers reporting multiple OATPs, RIF dose response, single RIF concentrations that were well above the reported EC50 for PXR-mediated CYP3A4 induction in vitro, and direct comparisons with CYP3A4 and ABC transporters in the same experiment. Some investigators were able to leverage proteomics, which is important when considering pre- versus posttranslation changes after addition of inducer to hepatocytes (Schaefer et al., 2012; Alam et al., 2018).
Summary of literature reports describing OATP expression profiling after addition of inducer to cultured human primary hepatocytes and human liver tissue slices in vitro
RIF and Phenobarbital.
As summarized in Fig. 1 and Table 4, it is evident that RIF and other compounds like phenobarbital are relatively weak inducers of OATP mRNA (vs. CYP3A4). The reports of Jigorel et al. (2006), Sahi et al. (2006), Chen et al. (2011), Badolo et al. (2015), Han et al. (2017), Moscovitz et al. (2018), and Niu et al. (2019) more or less indicate that OATP induction is only a fraction of that observed for CYP3A4; i.e., the relative Emax for OATP versus CYP3A4 (Emax ratio) at ∼10 µM RIF is ≤0.1. The report of Moscovitz et al. (2018) is noteworthy because the authors studied RIF over a concentration range (0.1–10 µM) and defined a “maximal observed induction” metric for OATP1B1 (3.1), OATP1B3 (0.5), OATP2B1 (2.9), and CYP3A4 (75). However, these data are based on mRNA expression, which does not necessarily translate to protein and function. Therefore, our attention also focused on the work of Schaefer et al. (2012) because the investigators deployed tandem liquid chromatography–mass spectrometry methods to quantitate transporter protein abundance in human hepatocytes after 48 hours with RIF (25 µM). In this instance, induction of CYP3A4 protein was evident (8.9-fold) when compared with OATP1B1, OATP1B3, OATP2B1, and the ABC transporters (Pgp, MRP2, and BCRP) that were quantitated (≤1.5-fold induction).
Amprenavir.
To date, Liu et al. (2012) have reported the highest magnitude induction of OATP1B1 mRNA in human hepatocytes (7-fold). This was achieved with the human immunodeficiency virus 1 protease inhibitor amprenavir, a known PXR agonist (Helsley et al., 2013). The same authors noted that this was accompanied by a 1.8-, 15-, 2.5-, and 1.5-fold increase in OATP1B3, CYP3A4, Pgp, and MRP2 mRNA expression, respectively. In the same study, RIF induced OATP1B1 mRNA expression 2.5-fold (Table 4). To our knowledge, there are no clinical reports describing tissue biopsy profiling after multiple doses of amprenavir. Therefore, such in vitro data also need to be corroborated.
CBZ.
As described above, Oscarson et al. (2006) reported ∼2-fold induction of OATP1B1 and OATP1B3 mRNA expression in the livers of two epileptic subjects receiving CBZ. Unfortunately, we are aware of only one report by Badolo et al. (2015) that described the lack of OATP1B1 mRNA induction in plated human hepatocytes after the addition of CBZ at a low concentration (5 µM). In the same study, the authors also showed that induction of cytochromes P450, BCRP, MRP2, and Pgp mRNA expression by CBZ was weak (≤2.0-fold). Because CBZ at high concentrations (>10 µM) is known to present as an inducer of CYP3A4 (Luo et al., 2002; Sugiyama et al., 2016), additional in vitro studies are warranted to further explore CBZ as an OATP inducer.
Precision-Cut Human Liver Tissue Slices
We felt it important to include the publication of Olinga et al. (2008) because the investigators used freshly prepared precision-cut human liver tissue slices and incubated with RIF (10 µM, 16 hours) and a low concentration of phenobarbital (50 µM, 24 hours). As expected, RIF induced CYP3A4 (14-fold), Pgp (4.8-fold), and MRP2 (2.5-fold) mRNA expression (Table 4). No induction of OATP1B3 (reported as OATP8) mRNA expression was observed (OATP1B1 and OATP2B1 not reported). On the other hand, the same authors did observe robust induction of OATP1B3 mRNA expression (5.6-fold) with phenobarbital, a CAR ligand, which accompanied mRNA increases for CYP3A4 (8.6-fold), MRP2 (12.4-fold), and Pgp (2.6-fold). Such marked induction of OATP1B3 has not been replicated by other investigators using cultured human hepatocytes incubated with higher phenobarbital concentrations (1 mM) (Schaefer et al., 2012). Unfortunately, we were unable to locate human liver biopsy expression data for phenobarbital-dosed subjects, and so the results reported by Olinga et al. (2008) cannot be corroborated.
Studies with FXR Agonists
For the sake of completeness, we also wanted to consider additional publications describing in vitro studies with agonists for additional nuclear receptors beyond PXR and CAR. A summary of our findings for a well-characterized FXR agonist (chenodeoxycholic acid) is tabulated (Table 5). Three reports described studies with conventional plated human primary hepatocytes (Meyer zu Schwabedissen et al., 2010; Liu et al., 2014; Krattinger et al., 2016), and one leveraged precision-cut human liver slices (Jung et al., 2007). In all cases, induction (≥3-fold) of bile salt export pump (BSEP, ABCB11) mRNA was evident and was indicative of FXR engagement. Although induction of OATP1B1 mRNA varied considerably across the studies (0.6- to 4-fold), the induction of OATP1B3 mRNA was more evident and in three of the four studies was reported as ≥3.5-fold (Table 5).
Chenodeoxycholic acid as inducer of OATP1B1, OATP1B3, MRP2, and BSEP in vitro
To date, chenodeoxycholic acid is the most robust and consistent in vitro inducer of any OATP (OATP1B3) reported. Unfortunately, we are not aware of any studies describing tissue biopsy expression profiling following chenodeoxycholate dosing, as was done for ursodeoxycholate (Marschall et al., 2005). Alternatively, we looked to a semisynthetic FXR agonist like obeticholic acid (6α-ethyl-chenodeoxycholic acid), which similarly elicits robust induction (>5-fold) of BSEP and small heterodimer partner mRNA expression in sandwich-cultured human hepatocytes (Zhang et al., 2017). But unlike chenodeoxycholic acid, clinical data are available for multidose obeticholic acid showing that it has a minimal effect on the AUC of an orally dosed BCRP or OATP probe drug (rosuvastatin) (Edwards et al., 2017). Consistent with such clinical data, Ijssennagger et al. (2016) have reported that obeticholic acid (1 µM) elicits a relatively modest effect on OATP1B1 and OATP1B3 mRNA expression (vs. induction of ABCB11 mRNA) in precision-cut human liver slices.
Studies with Peroxisome Proliferator–Activated Receptor and LXRα Agonists
To our knowledge, Meyer zu Schwabedissen et al. (2010) are the only group to describe OATP induction in vitro with an LXRα agonist such as TO-901317. The authors incubated TO-901317 with plated human hepatocytes and showed that OATP1B1 mRNA is induced (∼2.5-fold) with a more muted effect (∼1.2-fold induction) on OATP1B3 mRNA expression. Likewise, Rogue et al. (2010, 2011) have assessed the mRNA expression signatures of primary human hepatocytes after the addition of peroxisome proliferator–activated receptor (PPAR)γ (NR1C3) agonists (e.g., troglitazone and rosiglitazone) and dual PPARα/γ (NR1C1/NR1C3) agonists (e.g., muraglitazar). Test compounds were assessed at relatively high concentrations (up to 40–300 µM), and none was shown to induced OATP1B1 or OATP1B3 mRNA expression. In fact, suppression of OATP1B3 mRNA expression was noted. Such results are consistent with those of Chen et al. (2011), who similarly reported no induction of OATP1B1 mRNA in freshly prepared cocultures of human primary hepatocytes incubated with WY14643 (100 µM, PPARα agonist). In all cases, investigators described robust induction (≥6-fold) of known PPAR-regulated genes such as adipose differentiation–related protein.
Assessing OATP Induction: An Evolving Toolkit
As summarized in Table 6, various approaches could be jointly used to investigate the induction of OATPs in vitro and in vivo, support in vitro–to–in vivo extrapolations, and enable modeling and simulation exercises. However, many of the described tools need to be adapted to support the assessment of OATP induction by test compounds and still require validation prior to being widely accepted.
Summary of tools that could be available to assess the induction of human OATPs
In Vitro
Given the examples described in Tables 4 and 5, induction studies with human primary hepatocytes or tissue slices are advisable as primary in vitro screens. If OATP induction is evident (e.g., >2-fold increase in mRNA and protein) it should be compared with CYP3A4 and various ABC transporters and relative Emax values generated. Because new chemical entities may present unique induction signatures in hepatocytes, versus standard inducers such as RIF, CBZ, and phenytoin, additional studies with nuclear receptor transactivation assays (e.g., PXR, CAR, FXR, LXRα, vitamin D receptor) may have to be considered to rationalize the gene induction signatures observed in primary calls (Meyer zu Schwabedissen et al., 2010; Howe et al., 2011; Eloranta et al., 2012). In vitro induction studies could be extended to include burgeoning novel plate‐based and chip‐based microphysiological systems (e.g., liver organoids and liver-on-a-chip) that might better support the assessment of OATP expression and function and their modulation by test compounds (Ishida, 2018). To date, however, we are not aware of any reports describing the impact of test compounds on OATP expression in such systems.
Animal Models
Beyond in vitro assays, various attempts have been made to assess in vivo induction in extensively humanized mice or chimeric mice with humanized livers (Kakuni et al., 2013; Henderson et al., 2019). But in such instances, reports have largely focused on the induction of CYP3A4 by classic inducers such as RIF, and no data are available for OATP1B1 and OATP1B3. Evidently, there is a need to study the induction of human OATPs in such models.
In recent years, the cynomolgus monkey has increasingly been used as a model to investigate PXR-mediated induction of CYP3A by agents such as RIF. This is because monkey PXR is highly homologous to the human ortholog (96%) and cynomolgus monkey CYP3A is inducible by RIF both in vitro and in vivo (Kim et al., 2010; Li et al., 2014; Tahara et al., 2019). In fact, the RIF dose response curves for CYP3A4 induction in plated cynomolgus monkey and human primary hepatocytes are comparable (Kim et al., 2010). With this mind, it is not surprising that Niu et al. (2019) turned to the cynomolgus monkey as a model to study OATP induction by RIF. The authors described the dosing of cynomolgus monkeys with RIF for 7 days (18 mg/kg), using a protocol known to induce monkey CYP3A, and reported an ∼2-fold increase in antipyrine clearance (cytochrome P450 mediated) with no impact on pitavastatin, CPI, and CPIII plasma exposure. RIF similarly increases antipyrine clearance in human subjects (Bennett et al., 1982). The lack of OATP induction was confirmed after the addition of RIF (10 µM, 72 hour) to sandwich-cultured cynomolgus monkey hepatocytes; CYP3A4, MRP2, Pgp, BCRP, OATP1B1, and OATP1B3 mRNA was induced 58-, 2.2-, 1.4-, 1.4-, 0.9-, and 1.3-fold, respectively. Of note, no attempt was made by the authors to isolate liver or gut tissue, before and after the dosing of the monkeys with RIF, to support OATP, CYP3A4, and ABC transporter expression profiling.
Consistent with the report of Niu et al. (2019), mRNA expressing profiling of cynomolgus monkey liver tissue after ∼9 days of oral RIF (100 mg/kg), CBZ (80 mg/kg), or phenytoin (30 mg/kg) presents robust induction of various CYP3A forms (up to 19-fold) with no statistically significant changes in SLCO1B1 and SLCO1B3 expression (W. Hu and S. Arat, Pfizer Inc., unpublished results). Beyond a PXR agonist such as RIF, however, it is not known if the cynomolgus monkey is a suitable in vivo model to assess OATP induction via other mechanisms or nuclear receptors such as CAR, LXRα, FXR, or vitamin D receptor.
Liquid Biopsy Approaches to Facilitate Tissue Expression Profiling
As described above, the use of clinical drug probes and biomarkers to study OATP induction has its limitations, and human tissue biopsy expression profiling is likely the most direct approach to differentiate OATP induction versus CYP3A4 and ABC transporters. But because conventional tissue biopsy methods are not routine, researchers may wish to turn their attention to less invasive strategies such as plasma exosome–based liquid biopsy (Rodrigues and Rowland, 2019). For example, Rowland et al. (2019) recently described the assessment of CYP3A4 activity, protein, and mRNA induction using exosome preparations derived from the plasma of subjects who had received RIF (300 mg daily) for 7 days. With validation, a similar approach could be applied to the assessment of OATP protein and mRNA expression. Alternatively, the expression of OATPs in circulating human lymphocytes has been described, and attempts have been made to deploy them as liver and gut tissue surrogates; Yang et al. (2019) reported an ∼2-fold increase in OATP1B1 and BCRP mRNA expression in the circulating lymphocytes of subjects who had consumed an herbal medicine (2.5 g tanjin per day) for 7 days. Both the maximal plasma concentration (27%) and AUC (∼20%) of rosuvastatin were decreased in the same subjects. How such induction in circulating lymphocytes relates to OATP expression in tissue is presently not known. Most importantly, such results need to be confirmed, as others have failed to detect OATP1B1 mRNA expression in various human T-cell lines and blood mononuclear cells (Janneh et al., 2008).
Selective OATP Drug Probes and Biomarkers
From the standpoint of an OATP induction toolkit, perhaps the most challenging is the discovery and validation of clinical probe drugs and biomarkers that are highly selective for target gut and liver OATPs. In the absence of biopsy data, assessing and deconvoluting gut (e.g., OATP2B1) versus liver (e.g., OATP1B1 and OATP1B3) OATP induction will be difficult. Such a scenario is further complicated by coinduction of CYP3A4 and ABC transporters in both organs. As discussed above, changes in statin and OATP biomarker (CPI) PK cannot be necessarily ascribed to OATP induction in the absence of ABC transporter induction. To date, however, we are aware of only one study describing multidose RIF on plasma CPI levels (Kunze et al., 2018). Because CPI is selective for OATP1B1 and OATP1B3 (vs. OATP2B1) and limited to MRP2 as substrate (Bednarczyk and Boiselle, 2016; Kunze et al., 2018; Table 2), additional clinical studies evaluating its utility as an OATP induction trait measure are warranted. This is especially true for inducers that do not inhibit OATP or manifest autoinduction like RIF.
Concluding Remarks
Human OATPs (OATP1B1, OATP1B3, and OATP2B1) are important SLCs, and their modulation by inhibitory drugs has been shown to be of clinical relevance for many substrate drugs. When compared with such DDIs, however, there is less definitive clinical data describing their induction. Despite such limitations, it is possible to conclude that available literature describes only weak induction of OATPs (mostly mRNA data) following treatment with a gold standard potent PXR activator (RIF 600 mg once daily ≥7 days). By extension, it is assumed that the reported changes in individual statin PK (and OATP biomarker CPI plasma profiles), following multiple doses of RIF, can be ascribed to the induction of CYP3A4 and ABC transporters in gut and liver, as we all as RIF autoinduction.
Beyond RIF, disparate sets of data do provide some evidence that human OATP forms are inducible (>2-fold). As described above, OATP induction has been described for compounds such as atorvastatin (3-fold OATP2B1 mRNA induction in human liver biopsy samples), phenobarbital (∼6-fold OATP1B3 mRNA induction in human liver slices), CBZ (∼2-fold induction of OATP1B1 and OATP1B3 mRNA in livers of two epileptic subjects), chenodeoxycholate (∼2- to 6-fold OATP1B3 mRNA induction in plated human hepatocytes), TO-901317 (∼2.5-fold induction of OATP1B1 in plated human hepatocytes), and amprenavir (∼7-fold OATP1B1 induction in plated human hepatocytes). Although such results need to be corroborated, it appears that studies focused on the induction of gut and liver OATP1B1, OATP1B3, and OATP2B1 by agents other than RIF are warranted. Also, other OATP forms such as OATP1A2, OATP3A1, OATP4A1, and OATP4C1 should be considered (Eloranta et al., 2012; Oswald, 2019).
Evidently, the regulation of OATP expression and function is complex, and continuous vigilance will be needed, especially when attempting to model DDIs with new chemical entities that are OATP inducers in vitro or present as OATP substrates that may themselves become victims of induction. In reality, such data need to be balanced against a new chemical entity’s cytochrome P450 and ABC transporter substrate, induction, and inhibition signature. With substantial clinical evidence supporting Pgp induction for well-known CYP3A inducers (Lutz et al., 2018a,b; Supplemental Table 4), the US Food and Drug Administration has suggested leveraging information from CYP3A induction studies to rationalize the need for conducting additional clinical studies to evaluate the Pgp induction potential of an investigational drug using a known Pgp probe substrate (https://www.fda.gov/media/82734/download). On the basis of the exhaustive clinical and preclinical data reviewed here, however, a clear need to evaluate OATP induction potential in drug development does not emerge at this time. Most importantly, the apparent lack of OATP induction by drugs such as RIF in vivo should be diligently considered when developing extended PBPK models encompassing OATP induction-inhibition signatures in addition to the associated interaction mechanisms involving CYP3A4, CYP2C9, CYP2C8, MRP2, Pgp, and BCRP.
Acknowledgments
The authors would like to thank Mark West, Sarah Lazzaro, and Soraya Eatemadpour (Pfizer Inc.) for providing the transporter data presented in Supplemental Table 3. Drs. Wenyue Hu and Seda Arat (Pfizer Inc.) are also acknowledged for a personal communication regarding their unpublished cynomolgus monkey liver tissue mRNA expression profiling data following inducer administration. Nathaniel Johnson (Pfizer Inc.) is also acknowledged for a personal communication regarding the lack of induction of AADAC mRNA in human hepatocytes after the addition of RIF.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Rodrigues, Lai, Shen, Varma, Rowland, Oswald.
Footnotes
- Received October 14, 2019.
- Accepted December 6, 2019.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AADAC
- arylacetamide deacetylase
- ABC
- ATP-binding cassette
- AUC
- area under the plasma concentration versus time curve
- BCRP
- breast cancer resistance protein
- BSEP (ABCG2)
- bile salt export pump
- CAR (NR1I3)
- constitutive androstane receptor
- CBZ
- carbamazepine
- CPI
- coproporphyrin I
- CYP2C
- cytochrome P450 2C
- CYP3A4
- cytochrome P450 3A4
- DDI
- drug-drug interaction
- ECCS
- extended clearance classification system
- Emax
- maximal induction
- FXR (NR1H4)
- farnesoid X receptor
- LXRα (NR1H3)
- liver X receptor alpha
- MRP2 (ABCC2)
- multidrug resistance–associated protein 2
- OATP
- organic anion transporting polypeptide
- PBPK
- physiologically based pharmacokinetics
- Pgp (ABCB1)
- P-glycoprotein
- PK
- pharmacokinetics
- PPAR
- peroxisome proliferator–activated receptor
- PXR (NR1I2)
- pregnane X-receptor
- RIF
- rifampicin
- SLC
- solute carrier
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Perspective
- Regulation of OATP Expression and Function
- Interpreting and Modeling of Clinical DDI Data and the Challenge of Using RIF as a Model Inducer
- RIF as Inducer and Autoinducer
- OATP Induction in the Context of PK Modeling
- DDIs Involving Induction by RIF in the Context of ECCS
- Profiling of Human Tissue after Dosing with Inducer
- In Vitro Data
- Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI