


Abstract
Variability in drug pharmacokinetics is a major factor in defining drug efficacy and side effects. There remains an urgent need, particularly with the growing use of polypharmacy, to obtain more informative experimental data predicting clinical outcomes. Major species differences in multiplicity, substrate specificity, and regulation of enzymes from the cytochrome P450–dependent mono-oxygenase system play a critical role in drug metabolism. To develop an in vivo model for predicting human responses to drugs, we generated a mouse, where 31 P450 genes from the Cyp2c, Cyp2d, and Cyp3a gene families were exchanged for their relevant human counterparts. The model has been improved through additional humanization for the nuclear receptors constitutive androgen receptor and pregnane X receptor that control the expression of key drug metabolizing enzymes and transporters. In this most complex humanized mouse model reported to date, the cytochromes P450 function as predicted and we illustrate how these mice can be applied to predict drug-drug interactions in humans.
Introduction
Cytochrome P450-dependent mono-oxygenases (CYPs) play a major role in the metabolism and disposition of most therapeutic drugs, with >80% of currently used drugs metabolized by these enzymes (Williams et al., 2004; Guengerich, 2008). The P450 system comprises a number of multigene families, with individual members exhibiting a distinct pattern of substrate specificity (Nelson et al., 2004), and provides an adaptive response, where on exposure to drugs or environmental chemicals, transcription factors are activated, which increase the expression of specific cytochromes P450 and drug transporters, resulting in increased rates of elimination (Omiecinski et al., 2011). Two transcription factors, the constitutive androgen receptor (CAR) and the pregnane X receptor (PXR), play a pivotal role in this process (Stanley et al., 2006). This regulatory network is complex. Both CAR and PXR can be activated by the same compounds and can activate an overlapping spectrum of detoxication genes.
To define how drugs may be handled in humans, it is critical to obtain a detailed analysis of these pathways. A number of in vitro and in vivo approaches have been developed to predict how drugs interact with the human P450 system and the consequences for drug therapy involving in vitro screens using recombinant P450 enzymes, hepatic microsomal fractions, or isolated hepatocytes (Gebhardt et al., 2003) and in vivo pharmacokinetic studies in animals (Tang et al., 2007). These data are then extrapolated to the human situation using in silico algorithms (Rostami-Hodjegan, 2012; Tang et al., 2007).
Although valuable, these models have a number of limitations, including the challenge in predicting complex clinical outcomes from reductionist in vitro results and profound species differences between the pathways of drug disposition. In mice, for example, there are 34 cytochromes P450 in the major gene families involved in drug metabolism, i.e., the Cyp1a, Cyp2c, Cyp2d, and Cyp3a gene clusters, whereas in humans, there are eight (Nelson et al., 2004). Interestingly three human enzymes, CYP2C9, CYP2D6, and CYP3A4, account for ∼75% of all reactions, with CYP3A4 being the single most important human P450 accounting for ∼45% of phase 1 drug metabolism (Guengerich, 2008). Differences in the number of gene-duplication events during the past ∼65 million years since the human and mouse genomes have diverged prohibit the assignments of orthologous genes between humans and mice, and unsurprisingly, the substrate specificity of proteins within gene families varies greatly across species (Martignoni et al., 2006). Furthermore, regulation of cytochromes P450 and indeed drug transporters by xenobiotics through the nuclear receptors can also differ markedly between species (Scheer and Roland Wolf, 2013). This adds a further confounding factor in predicting in vivo drug pharmacokinetics in humans, restricting the utility of animal models in the prediction of drug-drug interactions (DDIs), which can lead to loss of efficacy and/or enhanced drug toxicity.
To improve the utility and predictability of in vivo models of drug metabolism, we and others have embarked on programs to humanize mice for both P450s and the transcription factors that control their expression (Cheung and Gonzalez, 2008; Shen et al., 2011; Scheer and Roland Wolf, 2013). These include humanized models for PXR (Xie et al., 2000; Zhou et al., 2006; Ma et al., 2007; Lichti-Kaiser and Staudinger, 2008; Scheer et al., 2008, 2010), CAR (Zhang et al., 2002; Scheer et al., 2008), CYP3A4 (Granvil et al., 2003; van Herwaarden et al., 2005, 2007; Yu et al., 2005; Cheung et al., 2006; Hasegawa et al., 2011; Kazuki et al., 2013), CYP2D6 (Corchero et al., 2001; Scheer et al., 2012b), and CYP2C9 (Scheer et al., 2012a). These models have provided valuable information on specific human drug responses in relation to pharmacokinetics, DDIs, and toxicity. Multiple humanized mouse models for PXR/CAR (Scheer et al., 2008), CYP2D6/CYP3A4 (Felmlee et al., 2008), PXR/CYP3A4 (Ma et al., 2008), and PXR/CAR/CYP3A4 (Hasegawa et al., 2011) have been created. Encouragingly, the latter model allowed quantitative predictions of clinical PXR/CYP3A4–mediated DDIs (Hasegawa et al., 2011). One limitation of the models created to date is that the human and remaining mouse proteins can both contribute to drug metabolism, as demonstrated by specific mouse Cyp2c enzymes being active in the metabolism of the CYP3A4 substrate midazolam (MDZ) (van Waterschoot et al., 2008). To minimize such problems, we report on the creation of a mouse model, in which mouse Pxr and Car, together with the Cyp2c, Cyp2d, and Cyp3a gene clusters, have been exchanged with human PXR, CAR, and the CYP2C9, CYP2D6, and CYP3A4/7 loci, respectively. This humanized mouse for PXR, CAR, CYP3A4/7, CYP2D6, and CYP2C9 (hPXR/CAR/CYP3A4/7/2D6/2C9) represents the most complex humanized model for any purpose described to date, and we demonstrate its functionality and utility in predicting pathways of drug metabolism and pharmacokinetics in humans.
Materials and Methods
Animal Husbandry
Mice were housed and maintained as described previously (Scheer et al., 2008). Mice were housed on sawdust in solid-bottom, polypropylene cages and provided with an RM1 pelleted diet (Special Diet Services Ltd., Essex, UK) and drinking water ad libitum before and throughout the studies. The temperature was maintained within the range of 19–23°C, and the relative humidity was within the range of 40–70%. A 12-hour light/dark cycle was maintained. hPXR/CAR/CYP3A4/7/2D6/2C9 were obtained from Taconic Biosciences GmbH (Cologne, Germany) and were used for studies at the University of Dundee following at least 5 days of acclimatization. Other transgenic strains were obtained from Taconic Biosciences GmbH and bred at the University of Dundee. C57BL/6 mice were used as wild-type (WT) controls. All animal procedures were performed under a United Kingdom Home Office license and approved by the Ethical Review Committee of the University of Dundee. Mice homozygous for all of the humanized genes were used for experimental studies.
The in vivo interaction of midazolam with ketoconazole was studied using 31–65 week old male hPXR/CAR/CYP3A4/7/2D6/2C9 mice. The effect of phenobarbital treatment on the Cyp2b expression in hPXR/CAR/CYP3A4/7/2D6/2C9 mice was investigated using 34–63 week old female animals. Wild-type and complex humanized mice used in all other studies were 18–22 week old males.
Generation of hPXR/CAR/CYP3A4/7/2D6/2C9 Mice
hPXR/CAR/CYP3A4/7/2D6/2C9 mice were generated from the previously described hPXR (Scheer et al., 2010), hCAR (Scheer et al., 2008), hCYP3A4/7 (Hasegawa et al., 2011), hCYP2D6 (Scheer et al., 2012b), and hCYP2C9 (Scheer et al., 2012a) mice by breeding. The Cyp3a locus in the hPXR/CAR/CYP3A4/7/2D6/2C9 model was further modified by the additional inactivation of mouse Cyp3a13, which was not deleted in the original hCYP3A4/7 model. This was achieved by deletion of a 4.3-kilobase BglII fragment, including exons 1 and 2, and the promoter region of Cyp3a13 essentially as described previously (van Herwaarden et al., 2007). Successful inactivation was demonstrated by the loss of Cyp3a13 mRNA expression in the liver and intestine of Cyp3a13−/− mice compared with WT controls (data not shown). hCYP3A4/7/Cyp3a13−/− mice were obtained by backcrossing hCYP3A4/7 and Cyp3a13−/− animals and the selection of offspring with allelic crossover of both loci.
Rifampicin and Phenobarbital Treatment
To induce the expression of CYP3A4, mice were given three i.p. daily doses of rifampicin (RIF) (10 mg/kg) or vehicle control (corn oil) and euthanized 24 or 48 hours after the last dosing using a rising concentration of CO2. In the case of phenobarbital (PB), mice were given three daily doses of PB (80 mg/kg × 2, and then 40 mg/kg i.p.) or vehicle control (phosphate-buffered saline) and euthanized 24 hours after the last treatment using a rising concentration of CO2.
Collection of Liver, Duodenum, Jejunum, and Ileum and Preparation of Microsomes
On the day of termination, the mice were weighed and the body weights were recorded, and then the mice were transferred to a suitable room for post mortem. The mice were killed by exposure to a rising concentration of CO2.
For liver collection and microsome preparation, the gall bladder was removed, and then the liver was removed, weighed, and scissor minced in ice-cold KCl (1.15% w/v) for subsequent liver subcellular fractionation. The fresh liver samples were homogenized in ice-cold SET buffer (0.25 M sucrose, 5 mM EDTA, and 20 mM Tris-HCL, pH 7.4) to make a 10% (w/v) homogenate solution (9 ml SET buffer/1 g liver) using the Polytron homogenizer (Kinematica AG, Lucerne, Switzerland). Microsomes were prepared by centrifugation first at 2000 rpm [Sorvall RTH-250 rotor; DuPont (UK) Limited Sorvall Products, Stevenage, UK] for 10 minutes at 4°C. Then, the supernatant was spun at 12,000 rpm (Sorvall SS-34 rotor, DuPont) for 20 minutes at 4°C. The resulting supernatant was centrifuged at 29,130 rpm (Sorvall TFT-45.6 rotor, DuPont) for 90 minutes at 4°C, and the microsomal pellets were resuspended in ice-cold SET buffer and stored at –70°C.
In the case of intestinal collections and microsome preparation, the small intestine was removed and flushed with ice-cold phosphate-buffered saline containing a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). The duodenum, jejunum, and ileum sections (approximately 10 cm each) were transferred into separate polypropylene tubes, flash frozen immediately in liquid nitrogen, and stored at approximately –70°C prior to use for microsome preparation. The frozen duodenum, jejunum, and ileum were homogenized in 5.5 ml of SET buffer containing the protease cocktail inhibitor (Roche) and phenylmethanesulfonyl fluoride (1 mM) using a Polytron homogenizer. Tissue homogenates were subjected to subcellular fractionation by differential centrifugation similar to that described for the preparation of liver microsomes. Microsomal fractions were stored at approximately −70°C prior to analysis.
Western Blotting
Detection of cytochromes P450 in microsomal fractions was carried out by SDS-PAGE and Western blotting.
CYP3A4.
A rabbit whole serum (WB-3A4; catalog #458234; BD-Gentest, Woburn, MA) was used to detect human CYP3A4. Three micrograms of microsomal protein were loaded per sample. The controls were liver microsomes from individual human donors with low (donor #HH13; catalog #452138; BD-Gentest) and high (donor #HFC205; catalog #452138; BD-Gentest) CYP3A4 activity, pooled human liver microsomes (catalog #452117; BD-Gentest), and membrane preparations from bacteria expressing human CYP3A4 recombinant protein (Pritchard et al., 1997) (0.1 pmol).
Cyp2b.
Cyp2b expression in liver microsomes (10 μg per sample) was visualized using rabbit polyclonal antibodies raised against rat CYP2B10 (Forrester et al., 1992). In-house prepared microsomes from naïve and PB-treated (80 mg/kg; three daily doses; i.p.) WT mice were used as controls.
CYP2C9.
Rabbit polyclonal antibodies against human CYP2C9 (catalog #Ab10317; Millipore, Temecula, CA) was used to detect human CYP2C9 in 10 μg of microsomal protein. Pooled human liver microsomes (10 μg; catalog #452117; BD-Gentest), in-house prepared microsomes from CYP2C9 humanized mice (10 μg) (Scheer et al., 2012a), and membrane preparations from bacteria expressing human CYP2C9 recombinant protein (Pritchard et al., 1997) (0.1 pmol) were used as controls.
CYP2D6.
CYP2D6 in microsomes (10 μg per sample) was detected using mouse monoclonal anti-human CYP2D6 antibodies (MAB-2D6; catalog #458246; BD-Gentest). The controls were liver microsomes from individual human donors with low (donor #HG43; catalog #452138; BD-Gentest) and high (donor #HH37; catalog #452138; BD-Gentest) CYP2D6 activity, pooled human liver microsomes (catalog #452117; BD-Gentest), and membrane preparations from bacteria expressing human CYP2D6 (Pritchard et al., 1998) recombinant protein (0.05 pmol).
Enzyme Activity Measurements and In Vitro Inhibition Studies
Midazolam 1′-Hydroxylation.
MDZ (0.39–50 μM) was incubated with liver microsomes [0.05 mg protein/ml for RIF-treated (termination was 24 hours after the last RIF dose) hPXR/CAR/CYP3A4/7/2D6/2C9 microsomes and 0.25 mg protein/ml for all other microsomal preparations] and NADPH (1 mM) in 50 mM HEPES buffer (pH 7.4) supplemented with MgCl2 (15 mM) and EDTA (0.1 mM) at 37°C. After 2 minutes, the reaction was stopped by adding to an equal volume of ice-cold acetonitrile containing the internal standard triazolam. Samples were centrifuged for 45 minutes at approximately 1500g on a Legend RT centrifuge (Sorvall, Newton, CT), and the concentration of 1′-hydroxymidazolam in the supernatant was measured by high-performance liquid chromatography–tandem mass spectrometry. Chromatographic separation was performed on a Luna C18(2) column (5 μm, 150 × 2.0 mm) (Phenomenex, Torrance, CA) using an injection volume of 10 μl and a run time of 16 minutes. The multiple reaction monitoring parameters for 1′-hydroxymidazolam and triazolam were 342.13, 343.08 (parent ion) and 203.05, 307.94 (collision ion), respectively. The concentrations of 1′- hydroxymidazolam were calculated from the calibration curve.
Inhibition of MDZ 1′-Hydroxylation by Ketoconazole.
Ketoconazole (0.01–0.5 µM) was added to the incubation mixtures, and the reactions were carried out essentially as described above, with the following exception. The protein concentration of liver microsomes from the RIF-treated (termination was 48 hours after the last rifampicin dose) hPXR/CAR/CYP3A4/7/2D6/2C9 mice was 0.25 mg protein/ml.
Inhibition of MDZ 1′-Hydroxylation by Erythromycin.
For mechanism-based inhibition, an erythromycin (0.152–3000 µM) microsome [final concentrations: 0.05 mg protein/ml for the RIF-treated (termination was 24 hours after the last RIF dose) hPXR/CAR/CYP3A4/7/2D6/2C9 microsomes and 0.25 mg protein/ml for all other microsomal preparations] mixture in 50 mM HEPES buffer (pH 7.4) supplemented with MgCl2 (15 mM) and EDTA (0.1 mM) was incubated at 37°C in a water bath for 5 minutes prior to the reaction start by the addition of an NADPH regenerating system (final concentrations: 1 mM NADPH, 4 mM glucose-6-phosphate, and 2 U/ml glucose-6-phosphate dehydrogenase). After 15 minutes, MDZ was added (final concentrations were 3.1, 4.46, and 4 µM for WT, human liver, and hPXR/CAR/CYP3A4/7/2D6/2C9 microsomes, respectively) and the reaction was terminated 2 minutes later by adding to an equal volume of ice-cold acetonitrile containing the internal standard triazolam. Samples were centrifuged for 45 minutes at approximately 1500g on a Legend RT centrifuge (Sorvall, Newton, CT), and the concentration of 1′-hydroxymidazolam in the supernatant was measured by high-performance liquid chromatography–tandem mass spectrometry as described in the section “Midazolam 1′-Hydroxylation.” The reversible inhibition experiment was performed as described above, except an NADPH regenerating system was added after the 15-minute incubation and subsequent addition of MDZ.
Inhibition of MDZ 1′-Hydroxylation by CYP3cide.
CYP3cide is a CYP3A4-specific time-dependent inhibitor (Walsky et al., 2012). A mixture of CYP3cide (0.5 and 5 µM), microsomes (0.8 mg protein/ml), and NADPH (1 mM) was incubated in 100 mM potassium phosphate buffer (pH 7.4) supplemented with MgCl2 (3.3 mM) at 37°C for 320 seconds before transfer of the reaction aliquot (10 times the final dilution) to the MDZ (final concentration of 50 µM) and NADPH (final concentration of 1 mM) solution in the phosphate buffer. After 2 minutes of incubation at 37°C, the MDZ hydroxylation was stopped by mixing with an equal volume of ice-cold acetonitrile containing the internal standard triazolam. No inhibitor controls were incubated with an equal volume of CYP3cide vehicle (ethanol). Positive controls contained ketoconazole (final concentration of 10 µM) in the MDZ reaction solution. Samples were centrifuged for 45 minutes at approximately 1500g on a Legend RT centrifuge (Sorvall, Newton, CT), and the concentration of 1′-hydroxymidazolam in the supernatant was measured by LC-MS/MS as described in the section “Midazolam 1′-Hydroxylation.”
Diclofenac 4′-Hydroxylation.
A diclofenac (4 μM) microsome (0.25 mg protein/ml) mixture with and without sulfaphenazole (50 µM) was incubated with NADPH (1 mM) in 100 mM potassium phosphate buffer (pH 7.4) supplemented with MgCl2 (3.3 mM) at 37°C. After 5 minutes, the reaction was stopped by adding to an equal volume of ice-cold acetonitrile containing the internal standard bufuralol. Samples were centrifuged for 45 minutes at approximately 1500g on a Legend RT centrifuge (Sorvall, Newton, CT), and the concentration of 4′-hydroxydiclofenac in the supernatant was determined by UPLC-MS/MS from the calibration curve. Compounds were separated on a Kinetex C18 column (1.7 μm, 50 × 2.1 mm) (Phenomenex) using an injection volume of 5 μl and a run time of 4 minutes. The multiple reaction monitoring parameters for 4′-hydroxydiclofenac and bufuralol were 312.15, 262.25 (parent ion) and 231.07, 188.13 (collision ion), respectively.
Bufuralol 1′-Hydroxylation.
A mixture of bufuralol (12.25 µM) and microsomes (0.25 mg protein/ml) with and without quinidine (5 µM) in a 100 mM potassium phosphate buffer (pH 7.4) supplemented with MgCl2 (3.3 mM) was incubated with NADPH (1 mM) at 37°C for 3 minutes before the reaction was stopped by mixing the reaction mixture aliquot with an equal volume of ice-cold acetonitrile containing triazolam as the internal standard. Samples were centrifuged for 45 minutes at approximately 1500g on a Legend RT centrifuge (Sorvall, Newton, CT), and the concentration of 1′-hydroxybufuralol in the supernatant was measured by UPLC-MS/MS. Chromatographic separation was performed on a Kinetex C18 column (1.7 μm, 50 × 2.1 mm) (Phenomenex) using an injection volume of 5 μl and a run time of 2 minutes. The multiple reaction monitoring parameters for 1′-hydroxybufuralol and triazolam were 278.43, 343.13 (parent ion) and 186.21, 308.10 (collision ion), respectively. Concentrations of 1′-hydroxybufuralol in the samples were calculated from the calibration curve.
Pentoxyresorufin O-Depentylation.
A mixture of pentoxyresorufin (5 µM) and microsomes (0.043–0.88 mg protein/ml) in 100 mM potassium phosphate buffer (pH 7.4) supplemented with MgCl2 (3.3 mM) was incubated at 37°C for 5 minutes before the reaction was initiated by injection of NADPH (final concentration: 1 mM). Generation of the fluorescent product was registered in a kinetic mode using Fluoroscan Ascent FL (excitation filter: 530 nm; emission filter: 585 nm; Labsystems, Helsinki, Finland). Slopes of the linear part of the kinetic curves were calculated using Ascent Software Version 2.4.1 (Labsystems). For each well with the reaction media, there was a control well containing the reaction mixture with resorufin (4 pmol). Before the addition of NADPH to the reaction wells, fluorescence was recorded both from the reaction and from the control wells. The average fluorescence was calculated and the difference between wells with and without resorufin was used for the conversion of the relative fluorescence units to the picomoles of the reaction product.
Absolute Protein Quantification of Human CYP Enzymes by LC-MC/MS
Absolute protein quantification of CYP2C9, CYP2D6, and CYP3A4 was carried out using microsomal fractions from intestinal and hepatic tissue by mass spectrometry–based targeted proteomics as recently described (Groer et al., 2014). GIFPLAER (CYP2C9), DIEVQGFR (CYP2D6), and EVTNFLR (CYP3A4) were used as validated protein-specific surrogate peptides in parallel with their stable isotope-labeled internal standards. Accuracy (analytical error) and precision (coefficient of variation) of the assay during the sample analysis were between ± 15%, respectively. Final protein expression data (pmol/mg) were calculated by normalization to the total protein content of the isolated microsomal fraction as determined by the bicinchoninic acid assay (Pierce, Rockford, IL). To compare the protein abundance in the humanized model relative to the levels in humans, healthy human liver tissue (n = 6, age: 34–68; gender: three males and three females) and the jejunum (n = 6, age: 51–73; gender: two males and four females) were analyzed in parallel.
In Vivo Midazolam Pharmacokinetics and DDI Study
Mice were divided into three groups (three animals per group). Two groups were given three daily doses of RIF (10 mg/kg; 10 ml/kg; i.p.), and one group received vehicle (corn oil; 10 ml/kg; i.p.). One of the RIF-treated groups was administered ketoconazole (KTZ) [20 mg/kg in polyethylene glycol (PEG) 400; 10 ml/kg; by mouth (PO)] 48 hours after the last RIF dose and the two other groups were given vehicle (PEG400; 10 ml/kg; PO). Midazolam (MDZ) (5 mg/kg in PEG400; 10 ml/kg; PO) was administered to all mice 30 minutes after the KTZ/vehicle dosing. Whole blood samples (10 μl) were collected at 10, 20, and 40 minutes and 1, 2, 3, 4, 6, 8, and 24 hours after the administration of MDZ, placed immediately into microfuge tubes containing a 10-μl heparin (15 U/ml) solution in MilliQ water, kept on ice, and stored at approximately −20°C prior to analysis. Concentrations of MDZ, 1′-hydroxymidazolam, 4-hydroxymidazolam, and KTZ in whole blood were measured by liquid chromatography–tandem mass spectrometry. Calibration standards were prepared in whole blood/water by adding an appropriate amount of corresponding analytical standards. The test samples and calibration standards (20 µl) were extracted in 65 μl of acetonitrile containing triazolam (0.05 μg/ml) as the internal standard, mixed on a Thermomixer compact (Eppendorf, Hamburg, Germany) for approximately 5 minutes, and centrifuged at approximately 16,100g for 10 minutes. The supernatant was transferred to a 96-well plate, and the concentrations of MDZ, 1′- hydroxymidazolam, 4-hydroxymidazolam, and KTZ were measured by ultra performance liquid chromatography–tandem mass spectrometry from the calibration curve. Chromatographic separation was performed on a Kinetex C18 column (2.6 μm, 50 × 2.1 mm) (Phenomenex) using an injection volume of 30 μl and a run time of 6 minutes. The multiple reaction monitoring parameters for MDZ, 1′-hydroxymidazolam, 4-hydroxymidazolam, KTZ, and triazolam were 326.08, 342.13, 342, 531.3, and 343.08 (parent ion) and 291.01, 203.05, 234, 82, and 307.94 (collision ion), respectively.
Data Analysis for the Inhibition of MDZ 1′-Hydroxylation by KTZ
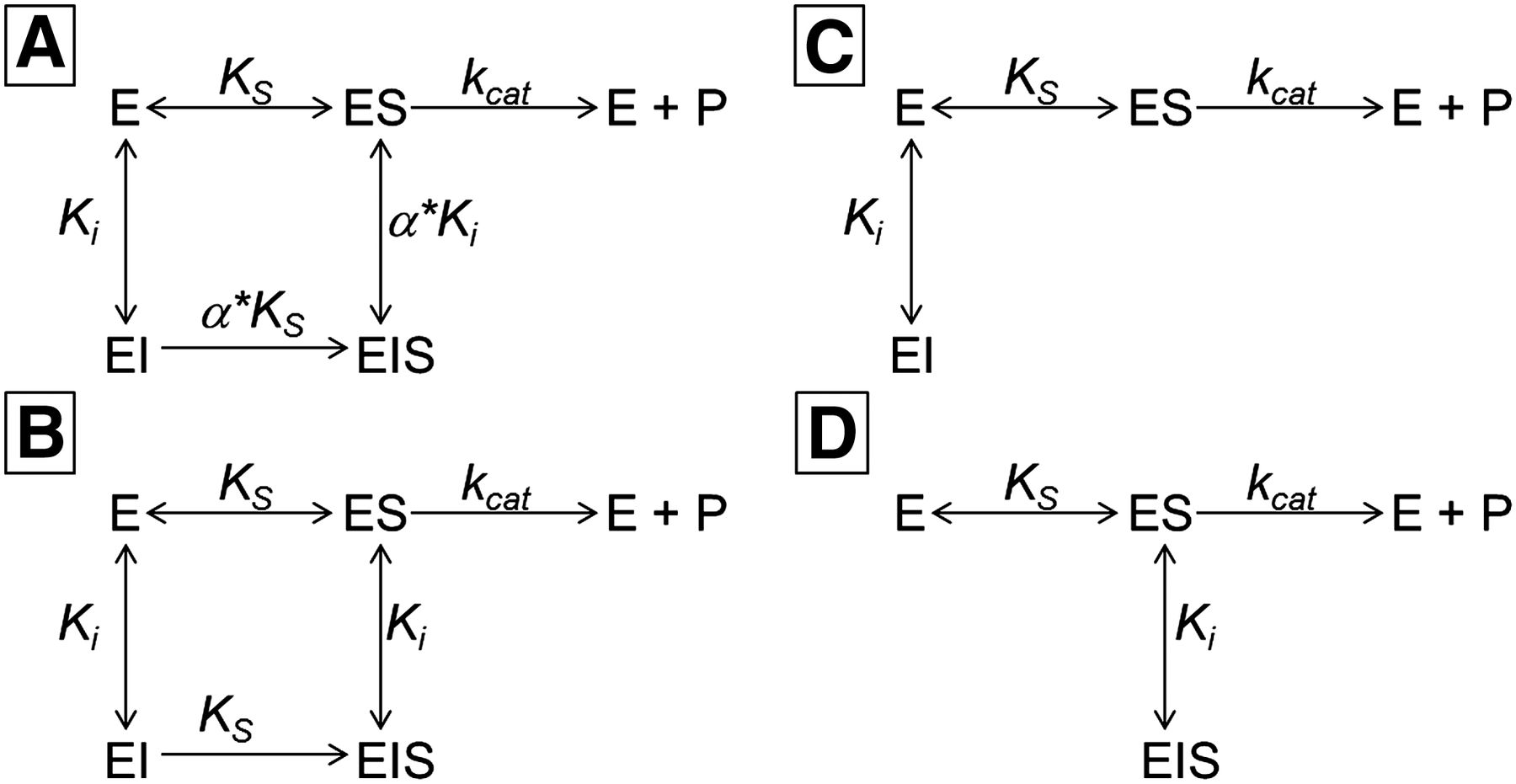
Data were analyzed by simultaneous nonlinear regression analysis (Kakkar et al., 1999, 2000) using GraFit 7.0.3 (Erithacus Software Limited, Horley, UK). For each microsomal preparation, a complete data set (with and without KTZ) was fit simultaneously using mixed, noncompetitive, competitive, and uncompetitive inhibition models (eqs. 1–4; Scheme 1).

Mixed (A), noncompetitive (B), competitive (C), and uncompetitive (D) inhibition models used in the data analysis of the inhibition of MDZ 1′-hydroxylation by KTZ.
MDZ and KTZ concentrations were two independent variables in the corresponding equations for simultaneous nonlinear regression.
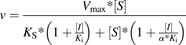 (1) Mixed inhibition
(1) Mixed inhibition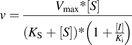 (2) Noncompetitive inhibition
(2) Noncompetitive inhibition (3) Competitive inhibition
(3) Competitive inhibition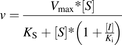 (4) Uncompetitive inhibition
(4) Uncompetitive inhibition where v is a reaction rate; Ks is a dissociation constant of the enzyme-substrate complex; Ki is a dissociation constant of the enzyme-inhibitor complex; Vmax is a reaction rate at the infinite substrate concentration in the absence of an inhibitor; α is a parameter describing the effect of inhibitor binding on the binding of the substrate and vice versa; and [S] and [I] are concentrations of the substrate and inhibitor, respectively.
For each microsomal preparation, fits produced by different inhibition models were compared using the QuickCalcs online application (GraphPad Software, San Diego, CA; http://graphpad.com/quickcalcs/aic1/). The preferable model among noncompetitive, competitive, and uncompetitive inhibition was selected by the lowest total sum of squares criteria. The selected model was subsequently compared with the mixed inhibition model by the F test.
In liver microsomes from WT mice, MDZ 1′-hydroxylation is catalyzed not only by cytochromes P450 from the Cyp3a subfamily, but also by mouse Cyp2c (Perloff et al., 2000). Mouse Cyp3a demonstrated a notably higher affinity to KTZ compared with mouse Cyp2c (Perloff et al., 2000). Accordingly, the kinetics of KTZ interaction with WT liver microsomes can be described in terms of inhibitor binding to two enzyme entities, which have different affinities to the compound. An enzyme unit with a relatively high affinity represents Cyp3a, and that with the lower affinity reflects Cyp2c (Scheme 2).

Two-enzyme inhibition model used in the data analysis of inhibition of MDZ 1′-hydroxylation by KTZ.
Substrate and inhibitor binding to the individual Cyp3a component was studied in microsomes from Cyp2c knockout (KO) mice, where the Cyp2c contribution to MDZ 1′-hydroxylation was effectively eliminated. The inhibition was described by a noncompetitive mechanism, with apparent dissociation constants of 2.6 and 0.013 µM for the enzyme complexes with MDZ and KTZ, respectively (eq. 5; Supplemental Table 1). Similarly, Cyp2c inhibition was investigated using Cyp3a knockout microsomes. The inhibition mechanism was also noncompetitive; however, the dissociation constants for MDZ (8.6 µM) and KTZ (1.2 µM) were markedly higher compared with those from the Cyp2c knockout microsomes (eq. 6; Supplemental Table 1).
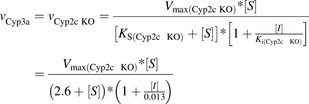 (5)
(5)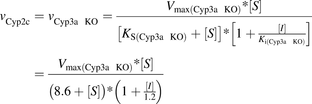 (6)
(6)where vCyp3a, (vCyp2c KO), and vCyp2c (vCyp3a KO) are the reaction rates catalyzed by the cytochromes P450 from the mouse Cyp3a and Cyp2c subfamily in microsomes from Cyp2c knockout and Cyp3a knockout mice, respectively; Vmax(Cyp2c KO) and Vmax(Cyp3a KO) are the reaction rates at the infinite substrate concentration in the absence of the inhibitor for Cyp2c knockout and Cyp3a knockout microsomes; Ks(Cyp2c KO) and Ks(Cyp3a KO) are enzyme-substrate complex dissociation constants for Cyp2c and Cyp3a knockout microsomes; Ki(Cyp2c KO) and Ki(Cyp3a KO) are enzyme-inhibitor complex dissociation constants; and [S] and [I] are substrate and inhibitor concentrations, respectively.
In WT microsomes, both Cyp3a and Cyp2c are expressed. Thus, the rate of MDZ 1′-hydroxylation can be presented as a sum of the reaction rates of the Cyp3a and Cyp2c components. It is reasonable to assume that KTZ and MDZ interactions with each enzyme component in the WT microsomes have similar dissociation constants (Ki and Ks) to those measured for the individual enzyme subfamilies in the corresponding knockout microsomes. Therefore, the dependency of the reaction rate in WT microsomes on the substrate and inhibitor concentrations can be expressed by eq. 7 (two-enzyme system):
 (7)
(7)where vWild type is the total reaction rate in WT microsomes; vCyp3a and vCyp2c are the reaction rates catalyzed by Cyp3a and Cyp2c enzymes, respectively; Vmax(Cyp3a) and Vmax(Cyp2c) are the reaction rates at the infinite substrate concentration in the absence of the inhibitor for Cyp3a and Cyp2c components; and [S] and [I] are the corresponding substrate and inhibitor concentrations.
The concentrations of the substrate and inhibitor in the equation are independent variables. Vmax(Cyp3a) and Vmax(Cyp2c) are parameters calculated by simultaneous nonlinear regression of the inhibition data, which show the impact of the corresponding cytochrome P450 subfamily on the total reaction rate. Using similar considerations, MDZ 1′-hydroxylation in microsomes from humanized mice for PXR, CAR, and CYP3A4/7 (hPXR/CAR/CYP3A4/7) can be presented as a general reaction catalyzed by human CYP3A4 and mouse Cyp2c components. Values of MDZ- and KTZ-binding parameters for each of the components were calculated using inhibition data from human and Cyp3a knockout liver microsomes, respectively (eqs. 1 and 6; Supplemental Methods; Supplemental Table 1). The corresponding reaction rate versus substrate and inhibitor concentration dependency is described by eq. 8.
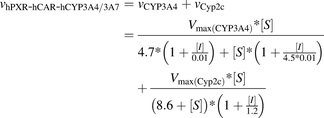 (8)
(8)where vhPXR-hCAR-hCYP3A4/3A7 is the total reaction rate in liver microsomes from hPXR/CAR/CYP3A4/7 mice; vCYP3A4 and vCyp2c are reaction rates catalyzed by CYP3A4 and Cyp2c enzymes, respectively; Vmax(CYP3A4) and Vmax(Cyp2c) are the reaction rates at the infinite substrate concentration in the absence of the inhibitor for the CYP3A4 and Cyp2c components; and [S] and [I] are the corresponding substrate and inhibitor concentrations.
MDZ- and KTZ-binding parameters obtained during inhibition studies with human and mouse Cyp3a/Cyp2c/Cyp2d triple knockout microsomes described the CYP3A4 and non-CYP3A4 components, respectively, in MDZ 1′-hydroxylation catalyzed by microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice (eq. 9).
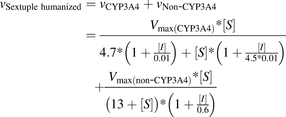 (9)
(9)where vSextuple humanized is the total reaction rate in liver microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice; vCYP3A4 and vNon-CYP3A4 are reaction rates catalyzed by CYP3A4 and non-CYP3A4 enzymes, respectively; Vmax(CYP3A4) and Vmax(non-CYP3A4) are reaction rates at the infinite substrate concentration in the absence of the inhibitor for CYP3A4 and non-CYP3A4 components; and [S] and [I] are the corresponding substrate and inhibitor concentrations.
Inhibition data generated in WT, hPXR/CAR/CYP3A4/7, and hPXR/CAR/CYP3A4/7/2D6/2C9 mouse liver microsomes were fitted with the two-enzyme model. The goodness of fit for the two-enzyme model and the most statistically preferable single enzyme model (mixed, noncompetitive, uncompetitive, or competitive) were compared using Akaike’s method (Supplemental Table 3).
Time-Dependent Inhibition of MDZ 1′-Hydroxylation by EMC
The published method of time-dependent inhibition data analysis using an IC50-shift experimental setup (Maurer et al., 2000; Berry and Zhao, 2008; Krippendorff et al., 2009) was modified to allow the simultaneous nonlinear regression of the IC50 curves. It was assumed that substrate metabolism follows Michaelis-Menten kinetics with the rapid equilibrium approximation, the inhibitor is competing with a substrate for the enzyme active site, and the enzyme-inhibitor complex can produce an inactive enzyme (Scheme 3).

Model of time-dependent enzyme inactivation in the presence of a substrate under the rapid equilibrium approximation Ks≈Km.
The dependency of the reaction rate of substrate metabolism on substrate and inhibitor concentration can be described by eq. 10
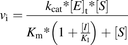 (10)
(10)where vi is the reaction rate in the presence of the inhibitor; kcat is the turnover number; [E]t is the active enzyme concentration at time t; [S] is the substrate concentration; Km is the Michaelis constant; [I] is the inhibitor concentration; and KI is the enzyme-inhibitor complex dissociation constant.
In the absence of the inhibitor, the reaction rate follows Michaelis-Menten kinetics and the active enzyme concentration [E]0 remains unchanged during the course of the reaction (eq. 11).
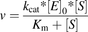 (11)
(11)where v is the reaction rate in the absence of inhibitor; and [E]0 is the active enzyme concentration.
The kinetics of enzyme inactivation in the presence of a mechanism-based inhibitor is described by eq. 12 (Mayhew et al., 2000).
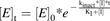 (12)
(12)where [E]t is the active enzyme concentration at time t; [E]0 is the active enzyme concentration at time 0; kinact is the rate constant of formation of the inactive enzyme; [I] is the concentration of the mechanism-based inhibitor; t is time; and KI is the enzyme-inhibitor complex dissociation constant.
After substitution of the active enzyme concentration [E]t in eq. 10, eq. 13 is obtained:
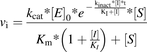 (13)
(13)In IC50 curves, the reaction rate is usually expressed as a percentage of that with no inhibitor. Therefore, eq. 13 can be divided by eq. 11 and multiplied by 100% (eq. 14).
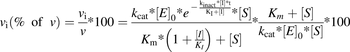 (14)
(14)where vi(% of v) is the reaction rate expressed as a percentage of that with no inhibitor. After some simplifications, eq. 15 can be obtained:
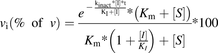 (15)
(15)If in the incubations, the substrate concentration was equal to Km, then further simplification of eq. 15 can be achieved (eq. 16)
 (16)
(16)The IC50 curves for the inhibition of MDZ 1′-hydroxylation by EMC obtained with and without preincubation with the inhibitor were analyzed by a simultaneous nonlinear regression analysis using eq. 15 or eq. 16 and the software GraFit 7.0.3 (Erithacus Software Limited, Horley, UK). The inhibitor concentration and time were independent variables, and kinact and KI were the calculated parameters.
Results
Generation and Phenotypic Analysis of hPXR/CAR/CYP3A4/7/2D6/2C9 Mice.
The generation of single hPXR (Scheer et al., 2010), hCAR (Scheer et al., 2008), hCYP3A4/7 (Hasegawa et al., 2011), hCYP2D6 (Scheer et al., 2012b), and hCYP2C9 (Scheer et al., 2012a) mice has been described previously. These mice have targeted the replacement of mouse Pxr and Car with the corresponding human genes and the mouse Cyp3a, Cyp2d, and Cyp2c gene clusters with human CYP3A4/7, CYP2D6, and CYP2C9, respectively. Human PXR and CAR in these models are known to be regulated by their corresponding mouse promoters; CYP3A4, CYP3A7, and CYP2D6 are known to be regulated by their cognate human promoters; and CYP2C9 is known to be regulated by the liver-specific mouse albumin promoter. Full genomic coding sequences of CAR, CYP3A4/7, and CYP2D6 are expressed in these models and hybrid human genomic/cDNA sequences of PXR and CYP2C9. The originally described hCYP3A4/7, hCYP2D6, and hCYP2C9 mice lack 14 of the 15 Cyp2c genes (Cyp2c29, Cyp2c37, Cyp2c38, Cyp2c39, Cyp2c40, Cyp2c50, Cyp2c54, Cyp2c55, Cyp2c65, Cyp2c66, Cyp2c67, Cyp2c68, Cyp2c69, and Cyp2c70), all nine Cyp2d genes (Cyp2d9, Cyp2d10, Cyp2d11, Cyp2d12, Cyp2d13, Cyp2d22, Cyp2d26, Cyp2d34, and Cyp2d40) and seven of the eight Cyp3a genes (Cyp3a11, Cyp3a16, Cyp3a25, Cyp3a41, Cyp3a44, Cyp3a57, and Cyp3a59), respectively. The mouse Cyp2c44 and Cyp3a13 genes, which are both located at some distance from the main clusters, were not deleted in these models. In the new model, we have additionally inactivated Cyp3a13 as described in Materials and Methods to eliminate a potential contribution of this P450 to drug pharmacokinetics. Cyp2c44, which has a low sequence homology with other mouse Cyp2c members, is not inducible by typical Cyp2c inducers and its role in drug metabolism is poorly defined (DeLozier et al., 2004), was not deleted. The individual humanizations included in the hPXR/CAR/CYP3A4/7/2D6/2C9 model are summarized in Fig. 1. Homozygous hPXR/CAR/CYP3A4/7/2D6/2C9 mice were generated from the single humanized mice by breeding.

Schematic overview of the individual humanizations included in the hPXR/CAR/CYP3A4/7/2D6/2C9 model. The intron/exon structure is shown for PXR and CAR, with the mouse exons indicated in black and with small type and human exons indicated in gray and with capitals. The striped boxes represent poly-adenylation motifs included in the targeting vectors. The structure of the gene clusters is illustrated in the case of CYP3A, CYP2D, and CYP2C, and genomic distances are indicated as kilobase (kb) or megabase (mb). Mouse genes and the direction of their transcription are shown as black arrows, and human genes are shown as gray boxes. Promoters driving the expression of the human genes are represented as white boxes. Dotted lines indicate the sites of replacement of the mouse with human sequences. For the sake of clarity, sequences are not drawn to scale.
Basal and Rifampicin-Induced Expression of CYP3A4, CYP2D6, and CYP2C9 Protein in hPXR/CAR/CYP3A4/7/2D6/2C9 Mice.
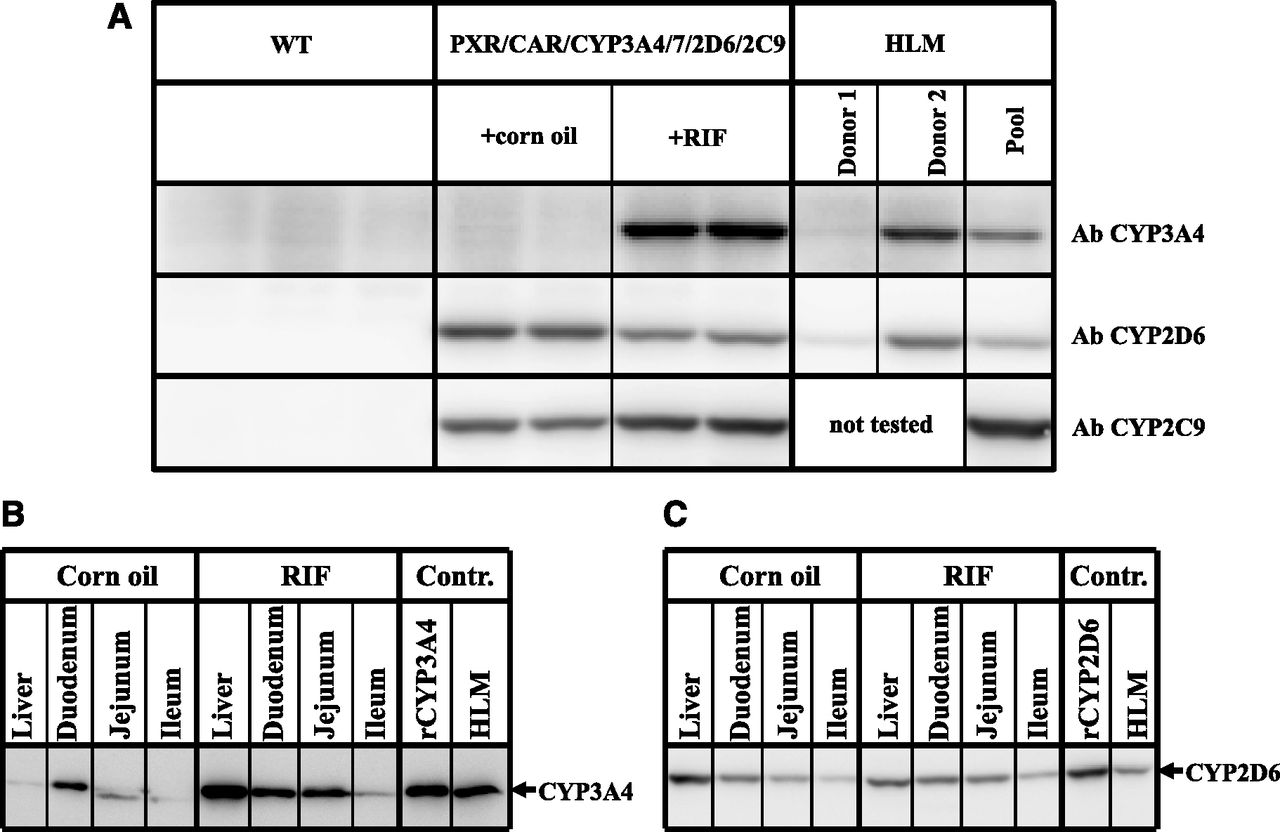
Western blot analysis showed that the constitutive hepatic expression of CYP2D6 and CYP2C9 was comparable to the average expression measured in human liver (Fig. 2A). Treatment with RIF slightly reduced the expression of CYP2D6 and slightly increased the expression of CYP2C9. As previously observed in the single humanized mouse model, the basal hepatic expression of CYP3A4 was low (Hasegawa et al., 2011); however, it was highly inducible following treatment with the human PXR activator RIF. At the dose used (10 mg/kg), hepatic CYP3A4 increased to levels equivalent to those found in human livers samples expressing high levels of this protein (Fig. 2A). The estimated concentration of CYP3A4 was determined using a CYP3A4 standard curve, and was 56 and 30 pmol/mg protein for the humanized mouse and the human liver sample, respectively (data not shown).
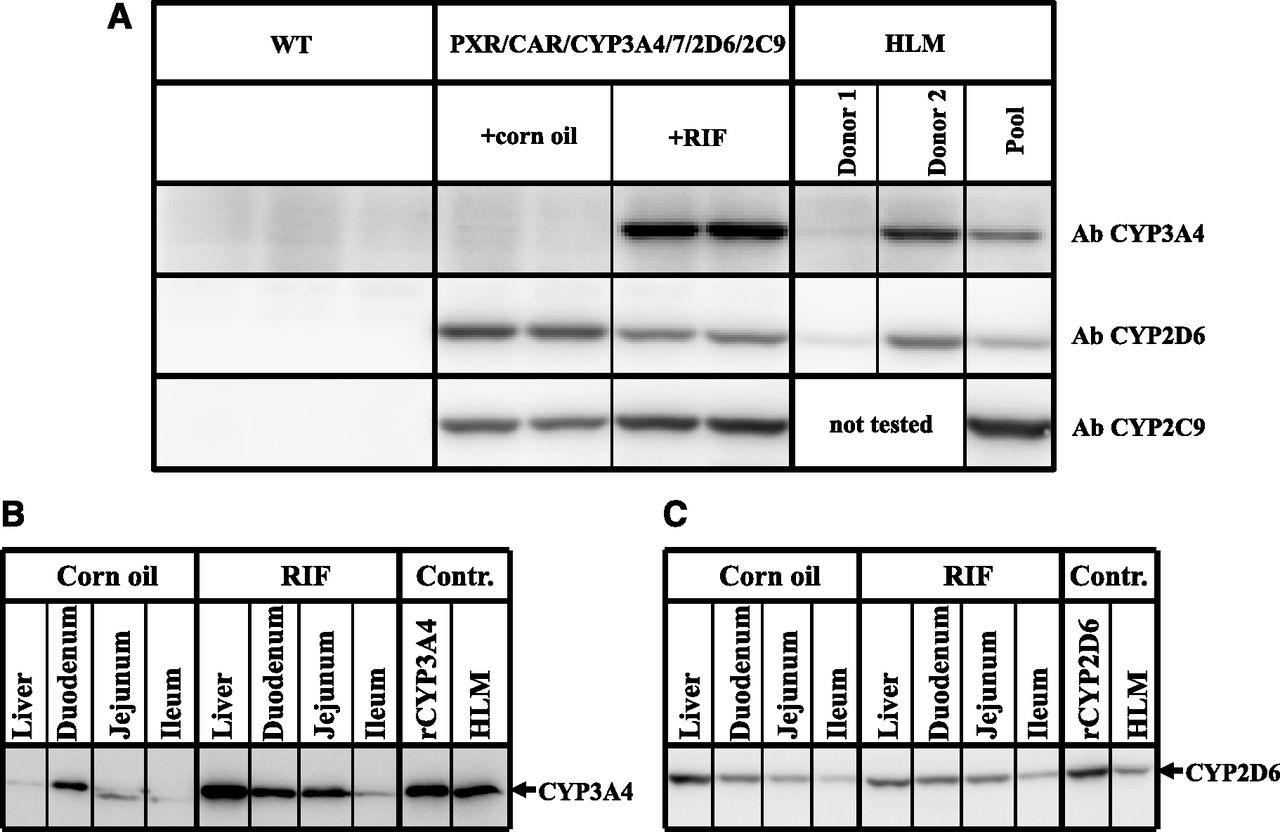
Basal and inducible expression of CYP3A4, CYP2D6, and CYP2C9 in hPXR/CAR/CYP3A4/7/2D6/2C9 mice. (A) Expression of CYP3A4, CYP2D6, and CYP2C9 protein in liver microsomes from WT mice (n = 3), corn oil–treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 2), RIF-treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 2), and human donors (HLM) determined by Western blot analysis using human specific antibodies (Ab). Donor 1 and donor 2 represent human individuals with low and high activity for CYP3A4 and CYP2D6, respectively. Donors with low and high activity for CYP2C9 were not available for this study and were therefore not tested. (B) CYP3A4 and (C) CYP2D6 protein expression in pooled liver, duodenum, jejunum, and ileum microsomes from corn oil– and RIF-treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 2 mice per treatment) determined by Western blot analysis. Controls (Contr.) are recombinant CYP3A4 or CYP2D6 and pooled HLM.
CYP3A4 and CYP2D6 expression was detectable constitutively in the intestine. In both cases, this was highest in the duodenum, followed by the jejunum and ileum (Fig. 2, B and C). RIF treatment significantly increased the CYP3A4 levels in all regions of the intestine, but had no effect on the expression of CYP2D6. As expected, CYP2C9, which is expressed under the control of the liver-specific albumin promoter, was not detected in the intestine of the multiple humanized model. This was determined by LC-MS/MS (see below).
Quantification of Hepatic and Intestinal CYP3A4, CYP2D6, and CYP2C9 Protein Levels in hPXR/CAR/CYP3A4/7/2D6/2C9 Mice by Mass Spectrometry.
Protein levels of CYP3A4, CYP2D6, and CYP2C9 were further quantified by LC-MS/MS in the liver, duodenum, and jejunum samples from control and RIF-treatedhPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 3 per group) and compared with the expression of these enzymes in the human liver and jejunum (n = 6 donors). It should be noted that the human liver samples used in this analysis were different to those used for the metabolism and Western blot studies. In agreement with the Western blot results, the expression of CYP3A4 in untreated mice was low but comparable with the expression level measured in the human donor with the lowest expression (1.3 pmol/mg protein) (Supplemental Table 1). Treatment of hPXR/CAR/CYP3A4/7/2D6/2C9 mice with RIF increased the hepatic CYP3A4 level to 220.7 ± 64.0 pmol/mg protein. This is approximately 4-fold higher than the level measured in a different set of samples by Western blot analysis (see above), but is within the range of that reported for the human liver (Watanabe et al., 2004; Ohtsuki et al., 2012; Liu et al., 2014). Consistent with the Western blot data, CYP3A4 was induced in the duodenum (from 2.8 ± 1.4 pmol/mg protein in control animals to 28.4 ± 18.3 pmol/mg protein in RIF-treated mice); however, because of the large variability in values between the samples, this change was not statistically significant. A comparison with the human duodenum expression was not possible as no human samples were available. In the jejunum, the constitutive level of CYP3A4 was similar to that found in human samples (3.6 ± 0.4 versus 2.1± 1.3 pmol/mg), but in this experiment, it was not increased on RIF treatment (3.1 ± 1.4 nmol/mg) .
As anticipated, treatment with RIF did not significantly change hepatic or intestinal CYP2D6 levels in the hPXR/CAR/CYP3A4/7/2D6/2C9 mice (Supplemental Fig. 1B; Supplemental Table 1). Compared with the average expression in the human livers tested, the CYP2D6 levels in these mice were ∼4-fold higher in the liver (7.0 ± 1.1 versus 27.2 ± 4.7 without and 28.3 ± 2.1 pmol/mg protein with RIF pretreatment) and 20- to 26-fold in the jejunum (0.2 ± 1.1 versus 4.8 ± 5.6 without and 3.7 ± 3.2 pmol/mg protein with RIF pretreatment), respectively.
Hepatic CYP2C9 expression was significantly increased from 12.3 ± 2.1 to 23.6 ± 2.3 pmol/mg protein in the hPXR/CAR/CYP3A4/7/2D6/2C9 mice by RIF, and both values were similar to the average expression of this enzyme in the human liver (18.8 ± 5.2 pmol/mg protein) (Supplemental Fig. 1C; Supplemental Table 1). Consistent with the use of the liver-specific albumin promoter to drive the expression of CYP2C9 in the transgenic mice, no CYP2C9 protein was detectable in the duodenum and jejunum of these animals compared with an average expression of 0.7 ± 0.3 pmol/mg protein in the human jejunum.
Assessment of CAR, CYP3A4, CYP2D6, and CYP2C9 Functional Activity.
Functional activity of PXR in the multiple humanized mouse model was demonstrated by the induction of CYP3A4 expression in response to treatment with the human PXR activator RIF (see above). To demonstrate functional CAR activity, the hPXR/CAR/CYP3A4/7/2D6/2C9 mice were treated with the CAR activator PB and induction of the Cyp2b10 protein and the Cyp2b-dependent pentoxyresorufin O-dealkylation was determined. The expression of Cyp2b10 protein in hPXR/CAR/CYP3A4/7/2D6/2C9 mice was induced to a level similar to that in WT mice treated with PB (Supplemental Fig. 2A). The Cyp2b10 induction by PB was significantly more pronounced than the PXR-mediated increase of Cyp2b10 by RIF. An ∼4- and ∼30-fold increase in the metabolism of the Cyp2b10 substrate pentoxyresorufin in RIF and PB treated samples, respectively, paralleled the observed changes in protein expression (Supplemental Fig. 2B).
CYP3A4 activity was measured using the probe substrate MDZ. Substrate dependencies for rates of MDZ 1′-hydroxylation were measured in corn oil WT–, corn oil–, and RIF-treatedhPXR/CAR/CYP3A4/7/2D6/2C9 mouse, and pooled human liver microsomes (Fig. 3A; Supplemental Table 2). The Vmax value of the microsomes from RIF-treated multiple humanized mice was approximately 3 and 10 times higher than that determined in pooled human and WT mouse liver microsomes, respectively. Furthermore, RIF treatment increased the Vmax of microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice by 58-fold compared with vehicle-treated controls, while the Km values were similar between the control and treated groups. The profound increase in MDZ 1′-hydroxylation in response to RIF is in agreement with the induction of CYP3A4 protein as determined by Western blot and protein quantification by LC-MS/MS (see above). These data demonstrate that CYP3A4 was functionally active in these mice. The similarity in Km values along with the observation of low constitutive CYP3A4 background protein expression (Fig. 2B; Supplemental Table 1) suggests that MDZ 1′-hydroxylation in corn oil–treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice is likely to be catalyzed by constitutively expressed CYP3A4.

Functional activity of CYP3A4, CYP2D6, and CYP2C9 in hPXR/CAR/CYP3A4/7/2D6/2C9 mice. (A) Substrate-concentration dependency for midazolam 1′-hydroxylation in microsomes from WT and hPXR/CAR/CYP3A4/7/2D6/2C9 mice and human donors. Formation of 1′-hydroxymidazolam was measured in pooled liver microsomes from WT mice (n = 3), a pool of human donors, and corn oil– and RIF-treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 2 per treatment). All measurements were performed in duplicates; symbols represent individual data; lines are nonlinear regression using the Michaelis-Menten kinetic model. (B) Bufuralol 1′-hydroxylation and (C) diclofenac 4-hydroxylation in liver microsomes from WT, hPXR/CAR/CYP3A4/7/2D6/2C9, Cyp2d, or Cyp2c knockout and Cyp2c/Cyp2d/Cyp3a knockout mice and human donors and inhibition of these reactions by quinidine (B) and sulfaphenazole (B). Data are expressed as mean ± S.D. (n = 3 for WT, Cyp2d, and Cyp2c knockout mice and human liver microsomes; n = 2 for the hPXR/CAR/CYP3A4/7/2D6/2C9 and Cyp2c/Cyp2d/Cyp3a knockout mice).
CYP2D6 activity was assessed by measuring metabolism of the probe substrate bufuralol. WT and hPXR/CAR/CYP3A4/7/2D6/2C9 liver microsomes showed a ∼14- and 8-fold higher rate of bufuralol 1′-hydroxylation than pooled human liver microsomes (HLMs), respectively (Fig. 3B). The higher activity in the multiple humanized model can be partially explained by the 4-fold higher expression of CYP2D6 protein. The reaction was almost completely inhibited by the CYP2D6-specific inhibitor quinidine in human liver microsomal samples and samples from hPXR/CAR/CYP3A4/7/2D6/2C9 mice. This was not the case in samples from WT animals, demonstrating a marked species difference in the metabolism of this compound. Interestingly, bufuralol 1′-hydroxylation in microsomes from WT mice was ∼11-fold higher than in microsomes from Cyp2d knockout mice(Scheer et al., 2012b), suggesting a major contribution from mouse Cyp2d enzymes. This difference increased to ∼230-fold when comparing the activity in WT mice to that in microsomes from Cyp2c/Cyp2d/Cyp3a triple gene cluster knockout mice (Scheer et al., 2014), indicating the additional involvement of mouse Cyp2c and/or Cyp3a enzymes in this reaction.
CYP2C9 activity was assessed by measuring the metabolism of the CYP2C9 probe substrate diclofenac. In agreement with the Western blot (Fig. 2A) and protein quantification (Supplemental Fig. 1C) data, diclofenac 4-hydroxylation in the liver microsomes from RIF-treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice was approximately 1.5-fold higher compared with vehicle-treated controls and ∼90% of that measured in the pooled HLMs (Fig. 3C). In contrast, this activity was 4.6-fold lower in microsomes from WT animals compared with the vehicle-treated multiple humanized mice. While the CYP2C9 specific inhibitor sulfaphenazole strongly decreased diclofenac 4-hydroxylation in human microsomes and microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice, it had no effect in liver microsomes from WT mice. Also, there was no difference in the activity between microsomes from WT, Cyp2c KO, and Cyp2c/Cyp2d/Cyp3a knockout mice, suggesting that none of these enzyme subfamilies catalyze this reaction in mice. The contribution of other mouse enzymes to this activity explains the retention of some diclofenac 4-hydroxylation activity in microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice in the presence of sulfaphenazole.
The above data demonstrate the functional activity of CAR, CYP3A4, CYP2D6, and CYP2C9 in the multiple humanized mice.
Assessment of CYP3A4 Contribution to 1′-Hydroxylation of Midazolam by Microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 Mice.
A limitation of single CYP3A4 humanized mice is the significant contribution of mouse Cyp2c enzymes to the metabolism of substrates, such as MDZ (van Waterschoot et al., 2008). This compromises the interpretation of any data obtained using this compound as a probe substrate in this model. The effect of the deletion of the Cyp2c gene cluster in the multiple humanized model on the metabolism of MDZ was investigated using liver microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice.
Whereas MDZ 1′-hydroxylation is catalyzed by Cyp3a and Cyp2c enzymes in WT mice, the Cyp3a component of this reaction is notably more sensitive to KTZ inhibition than the Cyp2c component (Perloff et al., 2000). We used this difference to delineate the contribution of CYP3A4/Cyp3a enzymes to MDZ 1′-hydroxylation from that of Cyp2c/non-CYP3A4 proteins in liver microsomes from WT and RIF-treated humanized PXR/CAR/CYP3A4/7 [hPXR/CAR/CYP3A4/7 (Hasegawa et al., 2011)] and hPXR/CAR/CYP3A4/7/2D6/2C9 mice. The data were analyzed by developing a two-enzyme kinetic model to describe the relative affinities and contribution of each enzyme to MDZ metabolism (see Materials and Methods and Supplemental Methods for details). Inhibition of MDZ 1′-hydroxylation in microsomes from WT, hPXR/CAR/CYP3A4/7, and hPXR/CAR/CYP3A4/7/2D6/2C9 mice by KTZ was analyzed by simultaneous nonlinear regression using the one-enzyme (mixed, noncompetitive, competitive, and uncompetitive) and corresponding two-enzyme inhibition models (eqs. 7–9 in Materials and Methods). The goodness of fit of the most statistically relevant one-enzyme model was compared with that by the two-enzyme model (Supplemental Fig. 3, A–C; Supplemental Table 3). Inhibition of MDZ 1′-hydroxylation by KTZ in WT and hPXR/CAR/CYP3A4/7 microsomes was significantly better described by the two-enzyme model than by the best fit one-enzyme model. This is consistent with the contribution of both Cyp3a and Cyp2c enzymes to this reaction in both sets of microsomes (Supplemental Table 3). The estimated contribution of hepatic Cyp2c in MDZ 1′-hydroxylation was ∼35% for both mouse lines. In contrast, when the analysis was carried out using data derived from the hPXR/CAR/CYP3A4/7/2D6/2C9 samples, the one-enzyme system produced the best fit (Supplemental Table 3), providing evidence that all of the metabolism can be ascribed to CYP3A4. Statistically, the most preferable mode of inhibition was noncompetitive, with calculated dissociation constants for the enzyme-substrate and enzyme-inhibitor complex of 4.1 and 0.047 μM, respectively.
To confirm the above findings, liver microsomes from WT, Cyp2c knockout, Cyp3a knockout, RIF-treated hPXR/CAR/CYP3A4/7, and hPXR/CAR/CYP3A4/7/2D6/2C9 mice were incubated with the potent and highly selective CYP3A4 time-dependent inhibitor CYP3cide (Walsky et al., 2012) prior to assessing MDZ 1′-hydroxylation activity. Recombinant CYP3A4 coexpressed with cytochrome P450 reductase in bacterial membranes rather than HLMs was used as HLM can contain CYP3A5, which also metabolizes midazolam but is CYP3cide insensitive. High concentrations of KTZ (10 μM) were used as a positive control in these studies. MDZ 1′-hydroxylation was not inhibited by CYP3cide in Cyp3a knockout microsomes, but was decreased by 79 and 84% in Cyp2c knockout microsomes after incubation with the 0.5- and 5-μM inhibitor. These data suggest that mouse Cyp3a, but not Cyp2c enzymes, are sensitive to CYP3cide (Fig. 4). The activity of liver microsomes from RIF-treated hPXR/CAR/CYP3A4/7 mice was inhibited by 67 and 81% by 0.5− and 5-μM CYP3cide, respectively. WT mouse liver microsomal fractions were more resistant to CYP3cide inhibition, being inhibited with 52 and 61%, respectively. This is consistent with the contribution of CYP3cide-insensitive mouse Cyp2c enzymes to MDZ 1′-hydroxylation. MDZ 1′-hydroxylation catalyzed by recombinant CYP3A4 was inhibited by 78 and 91%, and that by microsomes from the hPXR/CAR/CYP3A4/7/2D6/2C9 mice by 93 and 96% at 0.5− and 5-μM CYP3cide, respectively. The latter finding provided evidence that enzymes other than CYP3A4 play only a very minor role in MDZ 1′-hydroxylation in this model.

Inhibition of midazolam 1′-hydroxylation by CYP3cide in a range of liver microsomal samples. Formation of 1′-hydroxymidazolam was measured in pooled liver microsomes from WT (n = 3), Cyp2c knockout (n = 2), Cyp3a knockout (n = 2), and RIF-treated hPXR/CAR/CYP3A4/7 (n = 2) and hPXR/CAR/CYP3A4/7/2D6/2C9 mice (n = 2) as well as recombinant CYP3A4 coexpressed with cytochrome P450 reductase in bacterial membranes (rCYP3A4). Data are presented as mean ± S.D. (n = 3 measurements for each microsomal sample).
In addition to KTZ and CYP3cide, we tested the effect of the mechanism-based inhibitor erythromycin (EMC) on CYP3A4 on MDZ 1′-hydroxylation. This inhibitor is metabolized to a product that forms an inhibitory complex with CYP3A4 (McConn et al., 2004). Therefore, in one experiment, EMC, microsomes, MDZ, and NADPH were incubated concomitantly, while in the other, EMC was incubated with microsomes and cofactors for 15 minutes prior to the addition of MDZ (Supplemental Fig. 4, A–C; Table 1). When incubated concomitantly, an 18-fold lower IC50 was observed using HLM compared with WT mouse liver microsomes, indicating a marked species difference in the affinity to EMC. The IC50 in microsomes from hPXR/CAR/CYP3A4/7/2D6/2C9 mice was ∼8-fold lower compared with that measured in WT microsomes but ∼2-fold higher than that in pooled HLMs. The parameters for time-dependent inhibition were obtained by simultaneous nonlinear regression of the inhibition curves derived from experiments with and without preincubation with EMC as detailed in the Material and Methods. The estimated dissociation constant for the enzyme-inhibitor complex (KI) was also 21-fold higher in WT samples compared with pooled HLMs, which was only 3-fold lower than in microsomes from the multiple humanized mice. Calculated values for the rate constant of inactive enzyme formation (kinact) were similar between WT and pooled HLMs, but 3- to 4-fold higher in hPXR/CAR/CYP3A4/7/2D6/2C9 microsomes. EMC inactivation efficiency (kinact/KI) was 16-fold higher in pooled HLMs compared with WT samples, mostly due to the higher compound affinity of the human enzyme. The inactivation efficiency in microsomes from the multiple humanized mice was comparable to HLMs, and due to a combination of higher affinity to the inhibitor and a higher rate of inactivation, they were 21-fold higher compared with WT samples.
Kinetic parameters of midazolam 1′-hydroxylation inhibition by erythromycin in liver microsomes of different origin
In Vivo Interactions of Midazolam with Rifampicin and Ketoconazole in hPXR/CAR/CYP3A4/7/2D6/2C9 Mice.
Mice were treated i.p. with RIF (10 mg/kg) or vehicle for 3 days followed by a single PO dose of MDZ 48 hours post-RIF. One of the RIF-treated experimental groups was also given KTZ (PO) 30 minutes before MDZ administration. In the RIF-treated animals, MDZ exposure decreased almost 8-fold (Fig. 5, A and B), consistent with the observed strong induction of hepatic, and, to a lesser degree, intestinal CYP3A4 (Fig. 2B; Supplemental Fig. 1A). On coadministration of KTZ, a 1.9-fold increase in MDZ area under the curve (AUC) was measured (Fig. 5, A and B), which was indicative of inhibition of hepatic and/or intestinal CYP3A4. KTZ was rapidly eliminated, and within 3 hours post-MDZ, the KTZ total blood concentration was decreased by 90% (Fig. 5C).

Whole blood concentration versus time profiles of midazolam and ketoconazole in hPXR/CAR/CYP3A4/7/2D6/2C9 mice. (A) MDZ concentration versus time dependencies in hPXR/CAR/CYP3A4/7/2D6/2C9 mice treated with vehicle, RIF only, or RIF and KTZ. (B) MDZ area under the concentration versus time curve (AUC0–1440 min). (C) KTZ pharmacokinetics in hPXR/CAR/CYP3A4/7/2D6/2C9 mice. All data are expressed as mean ± S.D. (n = 3 mice per treatment group).
Discussion
The marked species differences between small mammals and humans limits their utility in predicting drug responses in humans. This includes predictions of pathways of drug disposition, bioavailability, drug efficacy, DDIs, and toxicity. Although useful, current in vitro systems and in silico modeling approaches often have limitations in predicting the complex DDIs in different tissues under clinical conditions. In this paper, we describe the generation and characterization of a novel sextuple humanized mouse model, in which the major mouse gene families involved in drug metabolism have been deleted and exchanged for their human counterparts. The generation of this model has involved the replacement of 33 mouse genes with six human genes and represents the most complex model of multiple humanizations for any biochemical pathway described to date.
We demonstrate the functionality of this model system and how it can be used to predict drug pharmacokinetics in humans. This versatile system can be used to investigate the relationship between drug exposure and efficacy, the capacity of drugs, at therapeutically relevant exposures to alter their own metabolism or of drugs given in complex combinations to cause DDIs. In the era of polypharmacy, this latter possibility addresses a current major area of clinical concern. An analysis of the use of prescribed drugs in Dundee, UK, for example, has shown that more than 40% of individuals over the age of 70 are taking more than eight drugs a day (2010 data; Medicines Monitoring Unit, http://www.dundee.ac.uk/memo/), demonstrating the enormous capacity for adverse drug reactions or loss of efficacy. Importantly, adverse drug reactions have been reported to be common causes of hospitalized death in the United States and UK (Lazarou et al., 1998; Pirmohamed et al., 2004). It is infeasible to evaluate all such DDIs by clinical trials, and the model described in this paper can support the evaluation of drug combinations that may be of concern. While possible DDIs are reasons for concern in many areas of clinical practice, including the treatment of cardiovascular and renal diseases, this is particularly important in oncology, where most anticancer drugs are still used at close to the maximum tolerated dose and changes in drug exposure can have life-threatening consequences.
The hPXR/CAR/CYP3A4/7/2D6/2C9 mice respond to drugs in a manner that more closely reflects the human situation both in terms of response to CAR and PXR activators and in relation to drug metabolism by key phase 1 metabolizing enzymes. When this model is used in conjunction with transgenic lines, where specific nuclear receptors or P450 gene clusters have been deleted alone or in combination, the ability to ascribe any results to a specific human P450 or signaling cascade is further increased (Scheer and Wolf, 2014). One significant advantage of the multiple relative to the single humanized mice stems from the significant functional redundancy in mouse cytochrome P450 gene families (van Waterschoot et al., 2008). This complicates the interpretation of experimental data, as demonstrated in this work and by others in studies with MDZ, which is oxidized by P450s expressed by both the Cyp2c and Cyp3a gene families (van Waterschoot et al., 2008). These gene families have both been deleted in the multiple humanized model, which circumvents this problem.
How the model overcomes species differences in drug inhibition is also reflected in the studies with the CYP2D6 and CYP2C9 substrates bufuralol and diclofenac. In WT mice, these activities are not significantly inhibited by the CYP2D6 inhibitor quinidine and the CYP2C9 inhibitor sulfaphenazole (Fig. 3, B and C), whereas in the humanized model, they are. A similar situation was observed for CYP3A4-mediated MDZ 1′-hydroxylation by EMC (Supplemental Fig. 4; Table 1). These data further illustrate the utility of these animals to overcome the confounding impact of species differences in drug metabolism.
The human P450s in the model described, with the exception of CYP2C9, are regulated by their own promoters, allowing the level of enzymes, such as CYP3A4, to be varied in the liver and/or the gut by prior treatment of the mice with a P450-inducing agent. This facilitates studies not only to establish the role of the P450 system in defining drug oral bioavailability, but also how variability in specific P450 levels will affect drug efficacy and/or side effects. For example, we demonstrate that pretreatment of the mice with RIF decreased the MDZ AUC ∼8-fold (Fig. 5, A and B). This is in good agreement with the results obtained from clinical studies (Backman et al., 1996). Also, the administration of KTZ to RIF-treated animals increased the MDZ AUC by 1.9-fold, which was somewhat lower than the ∼5-fold change reported in humans (Guest et al., 2011), and a possible explanation for this is as follows. At the time point of MDZ administration, the whole blood concentration of KTZ was ∼4 µg/ml. Taking the binding of MDZ to erythrocytes into account, this would translate to a plasma concentration of ∼6 µg/ml, i.e., the plasma level reported in patients (Daneshmend et al., 1981). However, in patients, this concentration reached a plateau at ∼2 hours, whereas in humanized mice, after 3 hours, it declined by 90% of the initial concentration. Furthermore, the KTZ dose of 20 mg/kg in our study can be extrapolated to a dose of 113 mg/70 kg in humans (Reagan-Shaw et al., 2008), which is lower than the 200 mg/kg KTZ dose used in the clinical studies that reported a 4.95–6.45 increase in MDZ AUC (Guest et al., 2011). Scaling the 1.9-fold MDZ AUC increase at a 113-mg equivalent KTZ dose in the multiple humanized mice to 200 mg would predict an ∼3.4-fold increase in exposure. The human to mouse dose translation described above does not account for the rate of drug elimination (Reagan-Shaw et al., 2008). Considering the strong induction of CYP3A4 activity in the RIF-treated hPXR/CAR/CYP3A4/7/2D6/2C9 mice and the relatively rapid elimination of the CYP3A4 substrate KTZ (Fitch et al., 2009) from the systemic circulation, the dose of the inhibitor in the induced hPXR/CAR/CYP3A4/7/2D6/2C9 mice has to be adjusted for the high activity of the major eliminating enzyme to adequately reflect the interaction of KTZ and MDZ in humans. An empirical method of the irinotecan dose calculation was developed to account for the interindividual variations of activities of CYP3A4 in humans, which required the collection of extensive in vivo experimental data (van der Bol et al., 2010). Applying a similar approach for dose translation between different species might be a more challenging task. Further dose adjustments of both RIF and KTZ may need to be made in future studies.
The hPXR/CAR/CYP3A4/7/2D6/2C9 mouse model described in this paper provides a powerful adjunct to existing experimental approaches for preclinical drug development and optimizing drug use in patients. It offers the potential to study complex in vivo DDIs involving the enzyme system responsible for ∼75% of the phase 1 metabolism of all marketed drugs. In addition, it provides an excellent basis for further development, for example, by introducing genetic modifications that reflect specific human diseases for predicting pharmacokinetic/pharmacodynamic relationships, and it has a significant potential application for the more informed design of clinical trials.
Acknowledgments
We wish to thank Julia Carr for technical assistance.
Authorship Contributions
Participated in research design: Scheer, Kapelyukh, Henderson, Oswald, Wolf.
Conducted experiments: Kapelyukh, Rode, Busch, McLaughlin, Lin.
Performed data analysis: Scheer, Kapelyukh, Oswald, Wolf.
Wrote or contributed to the writing of the manuscript: Scheer, Kapelyukh, Henderson, Oswald, Wolf.
Footnotes
- Received May 22, 2015.
- Accepted August 10, 2015.
↵1 Current affiliation: Independent Consultants, Cologne, Germany.
N.S. and Y.K. contributed equally to this work.
This work was supported in part by a Cancer Research UK programme grant [C4639/A10822] awarded to C.R.W. and by the InnoProfile-Transfer grant [Grant 03IPT612X] awarded to S.O. Part of this work was supported by ITI Life Sciences, Scotland.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- CAR
- constitutive androgen receptor
- CYP
- cytochrome P450
- DDI
- drug-drug interaction
- EMC
- erythromycin
- HLM
- human liver microsome
- hPXR/CAR/CYP3A4/7
- humanized mice for PXR, CAR, and CYP3A4/7
- hPXR/CAR/CYP3A4/7/2D6/2C9
- humanized mice for PXR, CAR, CYP3A4/7, CYP2D6, and CYP2C9
- KI
- enzyme-inhibitor complex dissociation constant
- kinact
- rate constant of inactive enzyme formation
- KO
- knockout
- KTZ
- ketoconazole
- MDZ
- midazolam
- PB
- phenobarbital
- PEG
- polyethylene glycol
- PO
- by mouth
- PXR
- pregnane X receptor
- RIF
- rifampicin
- WT
- wild type
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}