


Abstract
The aim of this study was to evaluate the contribution of metabolites to drug-drug interactions (DDI) using the inhibition of CYP2C19 and CYP3A4 by omeprazole and its metabolites as a model. Of the metabolites identified in vivo, 5-hydroxyomeprazole, 5′-O-desmethylomeprazole, omeprazole sulfone, and carboxyomeprazole had a metabolite to parent area under the plasma concentration–time curve (AUCm/AUCp) ratio ≥ 0.25 when either total or unbound concentrations were measured after a single 20-mg dose of omeprazole in a cocktail. All of the metabolites inhibited CYP2C19 and CYP3A4 reversibly. In addition omeprazole, omeprazole sulfone, and 5′-O-desmethylomeprazole were time dependent inhibitors (TDI) of CYP2C19, whereas omeprazole and 5′-O-desmethylomeprazole were found to be TDIs of CYP3A4. The in vitro inhibition constants and in vivo plasma concentrations were used to evaluate whether characterization of the metabolites affected DDI risk assessment. Identifying omeprazole as a TDI of both CYP2C19 and CYP3A4 was the most important factor in DDI risk assessment. Consideration of reversible inhibition by omeprazole and its metabolites would not identify DDI risk with CYP3A4, and with CYP2C19, reversible inhibition values would only identify DDI risk if the metabolites were included in the assessment. On the basis of inactivation data, CYP2C19 and CYP3A4 inhibition by omeprazole would be sufficient to identify risk, but metabolites were predicted to contribute 30–63% to the in vivo hepatic interactions. Therefore, consideration of metabolites may be important in quantitative predictions of in vivo DDIs. The results of this study show that, although metabolites contribute to in vivo DDIs, their relative abundance in circulation or logP values do not predict their contribution to in vivo DDI risk.
Introduction
Inhibitory drug-drug interactions (DDIs) can result in significant increases in the area under the plasma concentration–time curve (AUC) of an object drug by reducing systemic clearance or increasing bioavailability. Because of potential adverse effects exacerbated by inhibitory DDIs, they are of serious concern in drug development. Consequently, the ability to reliably identify potential in vivo inhibitors and predict the magnitude of DDIs from in vitro data is necessary. The recommended methods for performing preclinical risk assessment and quantitative DDI predictions have been outlined by the US Food and Drug Administration (FDA) (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf) and the European Medicines Agency (EMA) (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf). Included in the most recent FDA draft guidance is the recommendation that metabolites be considered in DDI risk assessment if metabolite AUC is greater than or equal to 25% of the parent AUC (AUCm/AUCp ≥ 0.25). The EMA further emphasizes that, if available, unbound concentrations should be used to determine relative exposures and that metabolites should have AUCm/AUCp>0.25 and represent >10% of total drug-related material.
With use of retrospective data, it has been recognized that many P450 inhibitors possess circulating metabolites (Isoherranen et al., 2009) and that inclusion of the metabolites in risk analysis can, in some cases, prevent false-negative predictions (Yeung et al., 2011). However, prospective studies aimed at understanding the importance of metabolites in DDI risk assessment are lacking, and the overall role of inhibitory metabolites in clinical DDIs and DDI predictions is still not well characterized. The relatively sparse data regarding inhibition potency of circulating metabolites (Yeung et al., 2011) has left the quantitative importance of metabolites in risk assessment to be controversial (Yu and Tweedie, 2013). In addition, very few studies have evaluated the importance of metabolites in irreversible interactions, despite the fact that most clinically important time dependent inhibitors (TDIs) possess circulating metabolites (VandenBrink and Isoherranen, 2010). Thus, more studies are needed to determine the role of circulating metabolites in reversible and irreversible P450 inhibition and to evaluate the correlation between abundance of metabolites in circulation and their contribution to inhibitory DDIs.
Omeprazole (OMP), which is metabolized by CYP2C19 and CYP3A4 (Andersson et al., 1994), is also an in vivo inhibitor of these two enzymes (Soons et al., 1992; Funck-Brentano et al., 1997; Yu et al., 2001; Angiolillo et al., 2011). OMP has been found to reversibly inhibit both CYP2C19 and CYP3A4 in vitro (Li et al., 2004; Zvyga et al., 2012), and recent investigations have shown that OMP is also a TDI of CYP2C19 (Ogilvie et al., 2011; Boulenc et al., 2012). Although in vivo DDIs with CYP2C19 substrates after OMP administration can largely be explained by CYP2C19 inactivation, the mechanisms of in vivo CYP3A4 interactions remain unexplained. It has been suggested that OMP metabolites may contribute to CYP2C19 inhibition (Ogilvie et al., 2011), but the metabolites have not been incorporated into DDI predictions and their circulating concentrations are not well characterized. In addition, it is possible that OMP metabolites are responsible for the in vivo CYP3A4 inhibition observed (Soons et al., 1992). Because OMP is a weak-to-moderate inhibitor of CYP2C19 and CYP3A4 in vivo, inclusion of metabolites in DDI risk assessment may change the risk categorization significantly. Two of the omeprazole metabolites, 5-hydroxyomeprazole (OH-OMP) and omeprazole sulfone (OMP-S), are known to be present in plasma after OMP administration, and OMP-S shows elimination rate–limited kinetics in vivo (Regårdh et al., 1990). Although OMP-S inhibits both CYP2C19 and CYP3A4 reversibly in vitro, its contribution to in vivo CYP2C19 and CYP3A4 inhibition was predicted to be insignificant, and inclusion of OMP-S to DDI risk assessment did not identify the in vivo CYP3A4 inhibition risk by OMP (Yeung et al., 2011). As such, OMP DDIs are insufficiently characterized, and further investigations are required to determine the potential contribution of the other circulating OMP metabolites to in vivo CYP2C19 and CYP3A4 interactions.
The aim of this study was to systematically evaluate the contribution of OMP metabolites to inhibitory DDIs observed with CYP2C19 and CYP3A4 after OMP administration. The circulating metabolites of OMP were identified in vivo in nine healthy human volunteers after a single 20-mg dose of OMP, and those metabolites meeting the recommended plasma exposure cutoff for DDI testing were evaluated in vitro for CYP2C19 and CYP3A4 inhibition. The obtained data were then used to assess the relative importance of the metabolites in in vivo DDIs and in DDI risk assessment.
Materials and Methods
Chemicals and Reagents.
Human liver microsomes (HLMs) from seven donors were obtained from the University of Washington Human Liver Bank maintained by the School of Pharmacy, University of Washington (Seattle, WA). HLMs were prepared using standard ultracentrifugation methods and were pooled before use. All donors were CYP2C19-extensive metabolizers and CYP3A5 nonexpressers. OMP, OH-OMP, 5′-O-desmethylomeprazole (DM-OMP), OMP-S, carboxyomeprazole (C-OMP), omeprazole sulphone N-oxide, omeprazole sulfide, omeprazole N-oxide, dextrorphan glucuronide, and 4-hydroxymephenytoin-d3 were purchased from Toronto Research Chemicals (Ontario, Canada). (S)-Mephenytoin, sulfatase from Aerobacter aerogenes, β-glucuronidase from Escherichia coli, and 4-nitrocatechol sulfate dipotassium salt were purchased from Sigma-Aldrich (St. Louis, MO). Midazolam, 1′-hydroxymidazolam, and α-hydroxymidazolam-d4 were purchased from Cerilliant (Round Rock, TX). Optima grade acetonitrile and water were purchased from Fisher Scientific (Waltham, MA). All other chemicals and general reagents were of analytical grade or better and were obtained from various commercial sources, such as Invitrogen (Carlsbad, CA) or Applied Biosystems (Foster City, CA).
Clinical Study.
The study protocol was approved by the University of Washington Institutional Review Board, and the study was registered at www.clinicaltrials.gov (NCT01361217). Plasma samples were obtained from the control day (no inhibitor) of the study. Nine subjects (five men and four women) participating in a cocktail study received a validated cocktail (Ryu et al., 2007) of 100 mg caffeine, 2 mg midazolam, 30 mg dextromethorphan, and 20 mg OMP (delayed release formulation) orally with 250 ml of water. It has been previously shown that the individual drugs in the cocktail do not affect the disposition of each other after a single-dose administration (Ryu et al., 2007). All participants were CYP3A5 nonexpressers and CYP2C19-extensive metabolizers based on genotype analysis. Blood samples were collected at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 12 hours after administration; plasma was separated from blood by centrifugation and stored at −80°C until analysis.
To establish the importance of conjugation reactions in the elimination of OMP metabolites, an aliquot of each plasma sample was treated with either β-glucuronidase from E. coli or sulfatase from A. aerogenes. For β-glucuronidase treatment, the enzyme was diluted in 100mM potassium phosphate buffer (pH 6.8) to a concentration of 500 units/ml; 50µl was then added to an equal volume of plasma, and the samples were incubated in the dark at 37°C overnight. Deconjugation of dextrorphan glucuronide in blank plasma was used as a positive control. For sulfatase treatment, the enzyme was diluted in 100mM potassium phosphate buffer (pH 7.1) to a concentration of 1 unit/ml; 50µl was added to an equal volume of plasma, and the samples were incubated in the dark at 37°C overnight. Deconjugation of 4-nitrocatechol sulfate dipotassium salt in plasma was used as a positive control. Deconjugation to 4- nitrocatechol was determined by measuring the absorbance at 515 nM. Negative control samples containing 50µl plasma and 50µl of the respective buffer were also incubated in the dark at 37°C overnight; 50µl of reaction mix was quenched in 100µl 1:3 acetonitrile to methanol containing 100nM OMP-d3 internal standard. OMP, DM-OMP, OMP-S, C-OMP, and OH-OMP were measured using liquid chromatography-tandem mass spectrometry (LC/MS/MS), as described below. The relative importance of the conjugates in plasma was determined by subtracting the concentrations of each analyte in control plasma from concentrations of the analytes in treated plasma.
Analysis of OMP and Its Metabolites in Human Plasma.
Plasma samples (67µl) were protein precipitated with 3:1 acetonitrile to methanol (133 µl) containing 100 nM d3-OMP and centrifuged twice at 3000g for 15 minutes, and the supernatant was transferred to clean plates between each spin. Samples were analyzed by LC-MS as described below. The AUC from time 0 to infinity (AUC0-∞) was calculated by the trapezoidal method using noncompartmental analysis and Phoenix software (Pharsight, Mountainview, CA).
LC/MS/MS Analysis of OMP and Its Metabolites in Human Plasma and 1′-Hydroxymidazolam and 4-Hydroxymephenytoin in HLM Incubations.
The concentrations of analytes in plasma samples and in incubations were determined using an LC/MS/MS system consisting of an AB-Sciex API 3200 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) coupled with an LC-20AD ultra-fast liquid chromatography system (Shimadzu Co., Kyoto, Japan). An Agilent ZORBAX XDB-C18 column (5μm; 2.1 × 50 mm) was used to separate 4-hydroxymephenytoin and 1′-hydroxymidazolam, and a Thermo Hypersil Gold 100 × 2.1 mm, 1.9 µm column (West Palm Beach, FL) was used to separate OMP, OH-OMP, DM-OMP, OMP-S, C-OMP, OMP N-oxide, OMP sulfone N-oxide, and OMP sulfide. The Turbo Ion Spray interface was operated in positive ion mode. A mobile phase of 0.1% aqueous formic acid (A) and acetonitrile (B) was used, and the injection volume was 10 µl. For the quantitative determination of 4-hydroxymephenytoin, a flow rate of 0.3 ml/min was used with a gradient elution starting from 5% B increased to 70% B by 3.0 minutes, then increased to 95% B by 3.1 minutes and kept at 95% B until 5.0 minutes, and returned to initial conditions by 7 minutes. The retention time of 4-hydroxymephenytoin was 4.0 minutes, and the mass transitions (m/z) were 235.157→150.200 and 238.157→150.200 for 4-hydroxymephenytoin and d3-4-hydroxymephenytoin, respectively. For the quantitative determination of 1′-hydroxymidazolam, OMP, OH-OMP, DM-OMP, OMP-S, C-OMP, OMP sulfone N-oxide, OMP sulfide, OMP N-oxide, and dextrorphan glucuronide, a flow rate of 0.4 ml/min was used with gradient elution with initial 10% of B increased to 90% B by 3.5 minutes and kept at 90% B until 5 minutes, then returned to initial conditions by 7 minutes. The mass transitions (m/z) and retention times were as follows: 342→324, 2.40 minutes (1′-hydroxymidazolam); 346→328, 2.87 minutes (α-hydroxymidazolam-d4); 346→198, 2.88 minutes (OMP); 349→198, 2.87 minutes (d3-OMP); 362→150, 2.99 minutes (OMP N-oxide); 362→214, 2.70 minutes (OH-OMP); 332→198, 2.48 minutes (DM-OMP); 362→150, 3.21 minutes (OMP-S); 376→149, 2.67 minutes (C-OMP); 330→182, 3.05 minutes (OMP sulfide); 378→166, 3.12 minutes (OMP sulfone N-oxide); and 434→258, 2.16 minute (dextrorphan glucuronide).
Analyst software, version 1.4 (AB Sciex), was used for data analysis. The day-to-day coefficient of variation percentage for all analytes was <15%. The limit of quantification for 1′-hydroxymidazolam, OMP, OH-OMP, DM-OMP, OMP-S, and C-OMP was 1 nM, and the limit of quantification for 4-hydroxymephenytoin was 50 nM.
Determination of Protein Binding of OMP and Its Metabolites and LogP Calculations.
OMP and its metabolites were added to 0.1 mg/ml HLM, 1.0 mg/ml HLM, or blank plasma to yield a final concentration of 1 µM (plasma) or 10 µM (HLM), and protein binding was determined using ultracentrifugation as described previously (Templeton et al., 2008; Lutz and Isoherranen, 2012). Samples were aliquoted into ultracentrifuge tubes (Beckman 343775) and incubated at 37°C for 90 minutes or spun at 435,000g at 37°C for 90 minutes with use of a Sorval Discovery M150 SE ultracentrifuge with a Thermo Scientific S100-AT3 rotor (Waltham, MA). The supernatant or the incubated sample was added to an equal volume of acetonitrile containing 100 nM d3-OMP, and samples were centrifuged at 3000g for 15 minutes at 4°C. HLM supernatant was transferred to a clean plate, and plasma supernatant was centrifuged a second time before analysis. The fraction unbound (fu) was calculated as the ratio of inhibitor concentration with or without ultracentrifugation. Calculated XlogP3 values were generated using Virtual Computational Chemistry Laboratory (vcclab.org).
Inhibition Experiments in HLMs to Determine IC50 Values.
Experiments were conducted at (S)-mephenytoin and midazolam concentrations of 20 μM and 1 μM, respectively [5-fold below reported Michaelis constant (Km) values]. Incubations were performed in solutions containing 100 mM potassium phosphate buffer (KPi; pH 7.4), and final incubation volumes were 100 µl and 150 µl for CYP2C19 and CYP3A4 assays, respectively. OMP or its metabolites (≥7 concentrations of 0.5–1000 µM), HLM (0.1 mg/ml), and substrate were preincubated for 10 minutes at 37°C before reactions were initiated by adding NADPH (1 mM, final concentration). Reactions were terminated after 20 minutes [(S)-mephenytoin] or 4 minutes (midazolam) by adding an equal volume of ice-cold acetonitrile containing 100 nM internal standard. All experiments were performed in triplicate. IC50 values were estimated by fitting Eq. 1 to the data with use of nonlinear least-squares analysis in GraphPad Prism (Graphpad Software, San Diego, CA) and the MULTI program (Yamaoka et al., 1981). Data are given as the mean of values obtained in at least three experiments with standard deviation.
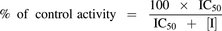 (1)
(1)IC50-Shift Experiments in HLMs for Time-dependent Inhibition.
Incubations were performed in solutions containing 100 mM potassium phosphate buffer (KPi; pH 7.4). For CYP2C19 studies, OMP and its metabolites (7 concentrations of 0.01–1000 μM) were incubated with 0.1 mg/ml HLM in 100 μl KPi buffer at 37°C for 30 minutes in the presence or absence of NADPH before (S)-mephenytoin (20 µM) or (S)-mephenytoin + NADPH were added. For CYP3A4 studies, OMP and its metabolites (7 concentrations of 0.1–1000 μM) were incubated in 150 μl KPi buffer with 0.1 mg/ml HLM at 37°C for 30 minutes in the presence or absence of NADPH before midazolam (1 µM) or midazolam plus NADPH were added. Reactions were terminated after 15 minutes [(S)-mephenytoin] or 3 minutes (midazolam) by adding an equal volume of acetonitrile containing 100 nM internal standard. The magnitude of the IC50 shift was determined from the ratio of the IC50 values after preincubation with and without NADPH. A shift ≥1.5 was considered to indicate irreversible inhibitory potential (Berry and Zhao, 2008; Grimm et al., 2009). Data are given as the mean of values obtained in triplicate experiments with standard deviation.
Inactivation Experiments in HLMs and CYP3A4 Supersomes to Determine Time Dependent Inhibition Values.
Mechanism-based inhibition was evaluated using the dilution method previously described (Waley, 1985). Incubations were performed in solutions containing 100 mM potassium phosphate buffer (KPi; pH 7.4). For CYP2C19 studies, HLMs (1 mg/ml) were incubated at 37°C with OMP, DM-OMP, or OMP-S (8 concentrations of 0.1–300 µM) and NADPH (1 mM). For CYP3A4 studies, HLMs (1 mg/ml) were incubated at 37°C with OMP or DM-OMP (7 concentrations of 1–500 µM) and NADPH (1 mM). At four designated time points, 10-μl aliquots were diluted 10-fold into activity assays containing a saturating concentration of (S)-mephenytoin (200 μM) or midazolam (30 μM; approximately 5-fold higher than substrate Km) and NADPH (1 mM final concentration). Reactions were allowed to proceed for 15 minutes [(S)-mephenytoin] or 3 minutes (midazolam) at 37°C. Inactivation experiments using CYP3A4 supersomes (b5 and P450 reductase coexpressed) were performed similarly to those described above with 5 pmoles CYP3A4. All reactions were terminated by adding an equal volume of acetonitrile containing 100 nM internal standard. Time dependent Inhibition kinetic parameters were determined by nonlinear least-squares analysis (Graphpad Prism or the MULTI program) by fitting Eq. 2 to the data:
 (2)
(2)where the λ is the apparent inactivation rate at a given inhibitor concentration, kinact is the maximum time-dependent inactivation rate (min−1), [I] is the inhibitor concentration and KI is the inhibitor concentration when the rate of inactivation reaches half of kinact (μM). Data are given as the mean of values obtained in triplicate experiments with S.D.
DDI Risk Assessment.
The DDI risk was determined by calculating the [I]/Ki ([I]/IC50) and λ/kdeg ratios for reversible and irreversible inhibition, respectively (Fujioka et al., 2012). In the present study, IC50 values can be regarded as equal to the Ki values because substrate concentrations were well below the relevant Km values. The value for in vivo λ was predicted using Eq. 2 by substituting the maximum concentration of inhibitor in plasma (Cmax) for in vivo inhibitor concentrations [I]. The values used for kdeg for CYP2C19, hepatic CYP3A4, and intestinal CYP3A4 were 0.00045 minute−1, 0.00032 minute−1, and 0.00048 minute−1, respectively (Fahmi et al., 2008; Nishiya et al., 2009; Ogilvie et al., 2011). The FDA recommends using total Cmax values for [I], whereas the EMA recommends using unbound Cmax values; thus, DDI risk was predicted using both values. Unbound IC50 and KI values were used in all calculations. Intestinal CYP3A4 inhibition was included in the risk assessment using an [I]g of dose/250 ml as the worst case scenario according to the FDA guidance. [I]/Ki ratios >0.1 and ≥0.02 were considered as indications of DDI risk, in accordance with the FDA and EMA guidance, respectively. DDI risk due to irreversible inhibition was considered as λ/kdeg values >0.1 and ≥0.25, as stated in the FDA and EMA guidance, respectively. The relative contribution of the metabolites was calculated as a fraction of the total predicted inhibition as described previously (Templeton et al., 2008).
Results
Identification of Significant OMP Metabolites.
Plasma samples were collected from nine subjects after a 20 mg single oral dose of OMP in a validated cocktail. The plasma samples were first analyzed to identify all OMP-related metabolites. A total of seven metabolites were identified (data not shown), and several additional OMP-related compounds, including conjugates of the metabolites, were detected, but the identity of all of these minor metabolites could not be determined. The circulating concentrations of OMP, OH-OMP, DM-OMP, OMP-S, C-OMP, OMP N-oxide, OMP sulfide, and OMP sulfone N-oxide were further measured using LC/MS/MS. The Cmax value of OMP was 660 nM. OMP N-oxide was detectable in 1–4 samples from each subject (18% of all samples), OMP sufide was detectable in 2–6 samples per subject (29% of all samples), and OMP sulfone N-oxide was detectable in 1–6 samples per subject (36% of all samples). However, all of the concentrations were below the lower limit of quantification of these compounds (63 nM for OMP N-oxide, 16 nM for OMP sulfide, and 31 nM for OMP sulfone N-oxide), making them minor circulating metabolites. The mean plasma concentration–time profiles of OMP, OH-OMP, DM-OMP, OMP-S, and C-OMP are shown in Fig. 1, and the Cmax, AUC0-∞, and unbound fraction in plasma (fu,p) are listed in Table 1. Treatment of plasma samples with β-glucuronidase and sulfatase led to an increase in DM-OMP plasma concentrations, but on the basis of quantification of the conjugated fraction, these conjugates were quantitatively minor. Similarly, deconjugation increased plasma concentrations of C-OMP and OH-OMP in some samples, but only slightly (< 20%), demonstrating that conjugates of these compounds are not quantitatively important in circulation. The total AUCm/AUCp ratios were ≥0.25 for OH-OMP, OMP-S, and C-OMP, but not for DM-OMP. On the other hand, unbound AUCm/AUCp ratios were ≥0.25 for OH-OMP, DM-OMP, and C-OMP, but not OMP-S, demonstrating a discrepancy between the two criteria. In addition, DM-OMP and OMP-S AUCs are likely to be <10% of total drug-related material, because OMP-S was 10% and DM-OMP was 1% of the total quantified drug-related material in this study. In the absence of radiolabeled data these values represent the upper limits of the abundance of the metabolites in relation to total drug-related material. Because all four of the circulating metabolites were found to have exposures above 25% that of OMP when either the total or unbound AUCs were considered (Table 1), they were all evaluated for in vitro inhibitory potential and included in in vivo risk assessment. However, it is unclear whether DM-OMP would be considered if OMP was a drug under development, because DM-OMP is a quantitatively minor metabolite.
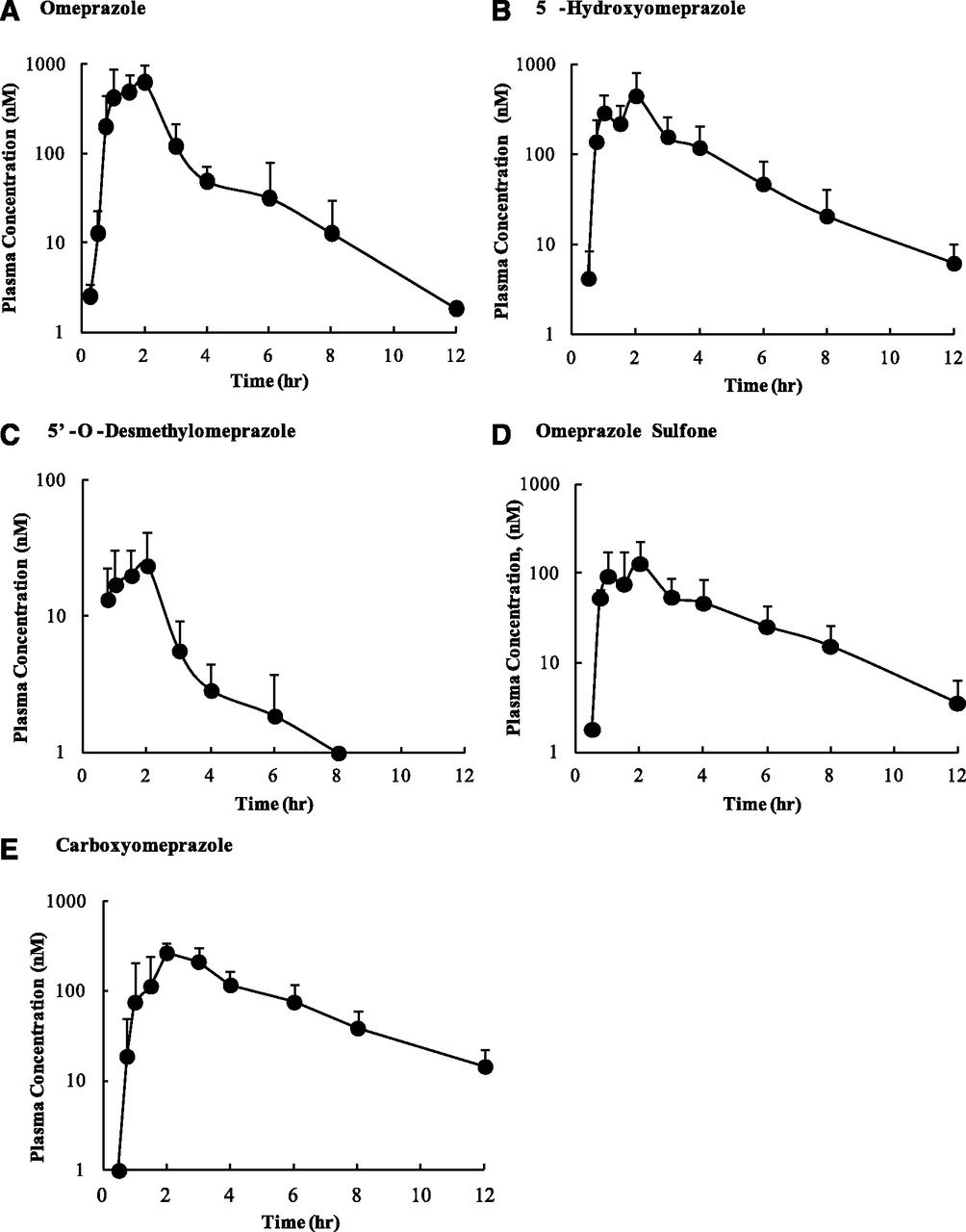
Mean plasma concentration–time profiles of omeprazole and its metabolites in nine healthy volunteers after oral administration of 20 mg omeprazole. Plasma concentrations of (A) omeprazole, (B) 5-hydroxyomeprazole, (C) 5′-O-desmethylomeprazole, (D) omeprazole sulfone, and (E) carboxyomeprazole were quantified. Data are shown as means ± S.D. (n = 9).
In vivo pharmacokinetic parameters of OMP and its metabolites
fu,p is the fraction of OMP and metabolites unbound in plasma, AUCm is the AUC of the metabolite, AUCp is the AUC of the parent (OMP), and AUCt is the summed AUC of all measured compounds.
CLogP values of OMP and its metabolites were calculated, and the values are listed in Table 1. With the exception of OMP-S, all metabolites were less lipophilic than OMP. When the logP values were compared with the fu,p for each compound, overall increased ClogP values were associated with increased plasma protein binding (decreased fraction unbound in plasma), with the exception of DM-OMP, which has a higher ClogP than either OH-OMP or C-OMP, but is less bound to plasma proteins (Table 1).
In Vitro Inhibition of CYP2C19 and CYP3A4 by OMP and Its Metabolites.
Reversible and irreversible inhibition of CYP2C19 and CYP3A4 by OMP and its metabolites was evaluated in pooled HLMs that were genotyped to be CYP2C19-extensive metabolizers and lack CYP3A5 expression. HLMs were selected as CYP2C19-extensive metabolizers for consistency and to obtain data not confounded by genetic variation in CYP2C19 expression levels. Similarly, HLMs genotyped as CYP3A5 expressers were excluded, because differentiation between CYP3A4 and CYP3A5 activity via selective substrates is not possible, and inactivation kinetics and inhibitory potency of omeprazole may be different between CYP3A4 and CYP3A5. OMP was found to be a more potent inhibitor of CYP2C19-catalyzed (S)-mephenytoin metabolism (IC50 value of 8.4 ± 0.6 μM) than CYP3A4-catalyzed midazolam hydroxylation (IC50 value of 40 ± 4 μM) (Supplemental Fig. S1; Table 2). All four metabolites (OH-OMP, DM-OMP, OMP-S, and C-OMP) also inhibited CYP2C19 and CYP3A4 reversibly, with OMP-S being the most potent inhibitor based on the reversible IC50 values (Supplemental Fig. S2; Table 2). Of interest, on the basis of the IC50 values, all of the metabolites except C-OMP were more potent CYP3A4 inhibitors than OMP. OMP-S was a more potent CYP2C19 inhibitor than were the other metabolites (Table 2). Although C-OMP also inhibited CYP2C19 and CYP3A4, the IC50 value for CYP2C19 could not be accurately determined because of lack of solubility of C-OMP (Supplemental Fig. S2; Table 2).
In vitro inhibitory parameters of OMP and metabolites for CYP2C19 and CYP3A4
N.D., not determined. Data are shown as means ± S.D. (n = 3).
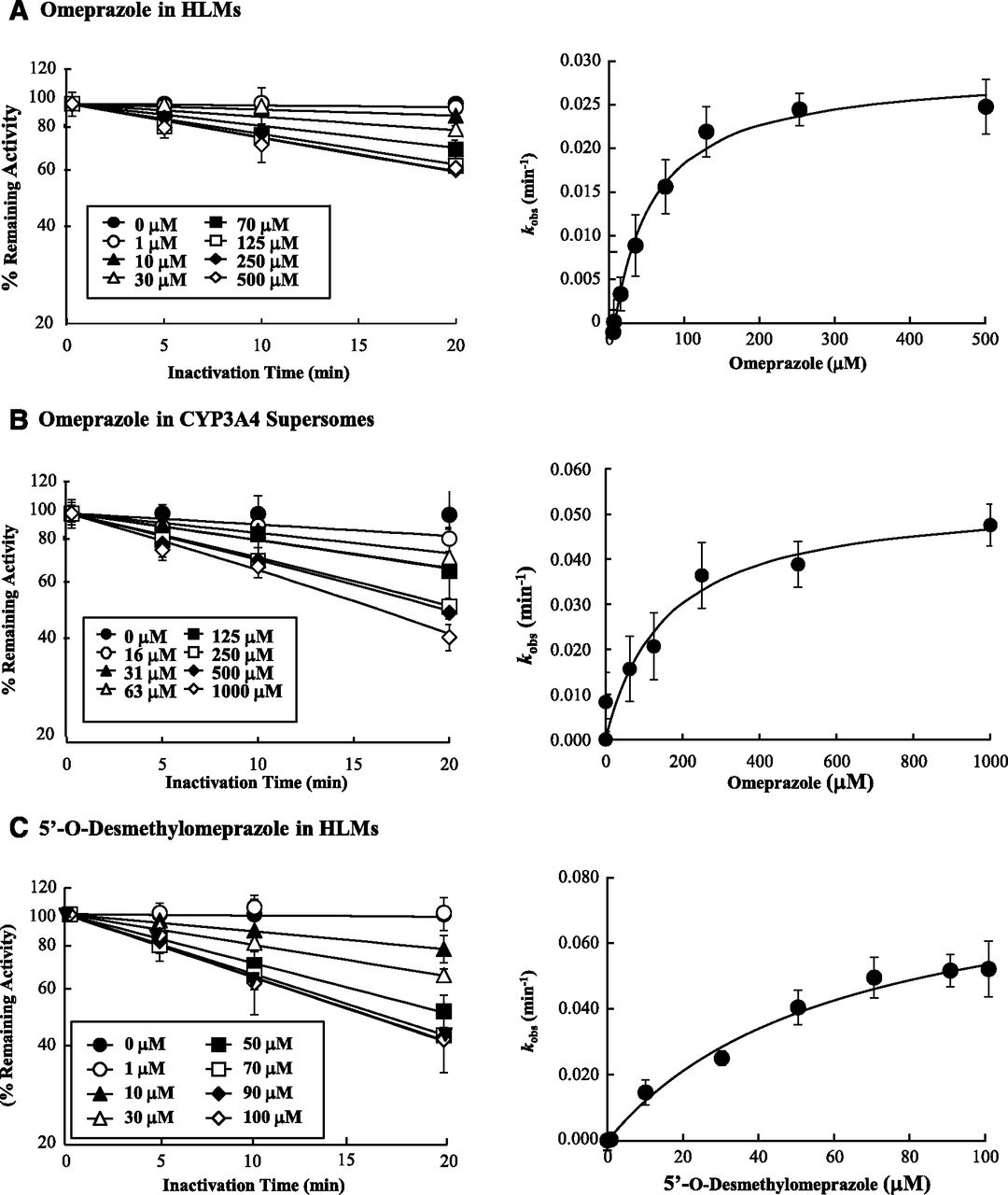
When IC50-shift experiments were performed, a 10-fold NADPH-dependent shift for CYP2C19 and 1.5-fold IC50 shift for CYP3A4 were observed with OMP, suggesting time-dependent inhibition of CYP2C19 and CYP3A4 by OMP (Fig. 2; Supplemental Table S1). Of the OMP metabolites, DM-OMP and OMP-S caused a ≥1.5-fold IC50 shift (7.3- and 2.1-fold, respectively) with CYP2C19, and DM-OMP also caused an IC50 shift (2.0-fold) with CYP3A4 (Supplemental Table S1), suggesting that DM-OMP and OMP-S may contribute to the inactivation of CYP2C19, and DM-OMP may contribute to CYP3A4 inactivation. No significant IC50 shift (<1.5-fold shift) was observed with either OH-OMP or C-OMP in the presence of NADPH with CYP2C19 or CYP3A4, suggesting that OH-OMP and C-OMP do not inactivate CYP2C19 or CYP3A4 (Supplemental Fig. S3; Supplemental Table S1).

NADPH-dependent IC50 shifts for OMP and its metabolites for CYP2C19-catalyzed (S)-mephenytoin hydroxylation and CYP3A4-catalyzed midazolam hydroxylation in HLMs. Inhibition of CYP2C19 and CYP3A4 by OMP (A and B), 5′-O-desmethylomeprazole (C and D), and OMP sulfone (E and F) is shown after a 30-minute preincubation with the inhibitor in the presence or absence of NADPH. All incubations were done as described in Materials and Methods. Data are shown as means ± S.D. (n = 3).
To characterize the time dependent inhibition of CYP2C19 and CYP3A4 by OMP and its metabolites, the KI and kinact values were determined for OMP and those metabolites that showed a significant IC50 shift. In agreement with the IC50 shift experiments, time- and concentration-dependent inhibition of CYP2C19 was observed with OMP, DM-OMP, and OMP-S (Fig. 3). All three compounds (OMP, OMP-S, and DM-OMP) showed similar TDI kinetics toward CYP2C19 with KI values of 5–9 μM and kinact values of 0.015–0.03 minute−1 (Table 2). Of the three CYP2C19 TDIs, OMP appeared to be most efficient, based on the kinact/KI ratios. Time- and concentration-dependent inhibition of CYP3A4 was also observed with OMP and DM-OMP, as predicted from the IC50 shift experiments (Fig. 4). OMP and DM-OMP were less potent inactivators of CYP3A4 than CYP2C19 based on the fact that their KI values were 7–10-fold higher toward CYP3A4. However, the kinact value for OMP was similar toward both CYP3A4 and CYP2C19, and DM-OMP had a slightly higher (4.3-fold) kinact value toward CYP3A4 than toward CYP2C19 (Table 2). In contrast to the rank order of inhibition efficiency between the compounds with CYP2C19, DM-OMP was a more efficient TDI of CYP3A4 than was OMP, based on the 3-fold higher kinact/KI ratio.

Time dependent inhibition kinetics of CYP2C19 by OMP (A), 5′-O-desmethylomeprazole (B), and OMP sulfone (C) in HLMs using (S)-mephenytoin hydroxylation as a probe. The left panels show the time-dependent inhibition of CYP2C19 at various concentrations of OMP and its metabolites. The right panels show the fit of Eq. 2 to the data. Data are shown as means ± S.D. (n = 3).

Time dependent inhibition kinetics of CYP3A4 by OMP in HLM (A), and CYP3A4 supersomes (B), and 5′-O-desmethylomeprazole in HLMs (C). The left panels show the time-dependent inhibition of CYP3A4 at various concentrations of OMP and DM-OMP. The right panels show the fit of Eq. 2 to the data. Data are shown as means ± S.D. (n = 3).
To assess whether CYP2C19-mediated formation of the DM-OMP is required for the time dependent inhibition of CYP3A4 by OMP in HLMs, inhibition of CYP3A4 by OMP was further evaluated using CYP3A4 supersomes. As shown in Fig. 4, time dependent inhibition of CYP3A4 by OMP was observed in the absence of CYP2C19, and OMP had a higher KI (157 μM) and kinact (0.054 minute−1) toward CYP3A4 in supersomes, compared with HLMs. The inhibition efficiency in supersomes was slightly lower than in HLMs, based on the kinact/KI ratio.
The ClogP of OMP and its metabolites were compared with the IC50 values and kinact/KI ratios to assess whether lipophilicity was predictive of the inhibition potency (Tables 1 and 2). A correlation analysis was not possible because of the small sample size. However, for CYP2C19, reversible inhibition potency appears to be higher for more lipophilic compounds. For CYP3A4, the ClogP rank order did not predict the rank order of inhibitory potency of the metabolites. To determine that the rank order of inhibitory potency and ClogP was not confounded by protein binding issues, the unbound fractions of OMP and its metabolites were determined in HLMs at the protein concentrations used for reversible and time-dependent inhibition experiments and are shown in Table 2. The nonspecific binding of OMP and its metabolites was insignificant at both HLM protein concentrations.
Contribution of OMP and Its Metabolites to DDI Risk Assessment.
With use of the in vitro inhibition parameters and in vivo concentrations of OMP, OH-OMP, DM-OMP, OMP-S, and C-OMP after the 20-mg single oral dose, the DDI risk and metabolite contribution to DDI risk assessment was evaluated. First, the DDI risk was predicted based on reversible [I]/Ki ratios for CYP2C19 and CYP3A4 (Table 3). Although a CYP2C19- or CYP3A4-mediated DDI risk was not identified on the basis of reversible [I]/Ki ratios of OMP with total and unbound Cmax values, inclusion of all the metabolites with total Cmax values did indicate a CYP2C19-mediated DDI risk as the sum of the [I]/Ki ratios was >0.1. However, use of the 0.02 cutoff with unbound Cmax value did not identify DDI risk. The CYP3A4-mediated DDI risk was missed both with total and with unbound Cmax values even when metabolites and gut inhibition were included in the risk assessment (Table 3).
Assessment of in vivo CYP2C19 and CYP3A4 inhibition risk by OMP and metabolites
Because substrate concentration was <Km for all IC50 experiments, the IC50 was assumed to be equivalent to Ki. [I]/IC50 and λ/kdeg values are calculated based on in vitro inhibitory parameters shown in Table 2. Either total or unbound Cmax were used for inhibitor concentration. N.D., not detected.
Identification of OMP as an irreversible inhibitor of both CYP2C19 and CYP3A4 was critical for appropriate risk assessment of in vivo DDIs. The λ/kdeg value for OMP was >0.1 and 0.25 with CYP2C19 using total or unbound Cmax values, respectively, and inclusion of the metabolites increased the overall DDI risk (Table 3). With CYP3A4, the λ/kdeg value for OMP identified the DDI risk when total Cmax value was used or gut inhibition was considered. With use of unbound Cmax values, a hepatic CYP3A4 inhibition risk was only detected using the λ/kdeg cutoff of 0.1 and when DM-OMP was included in the risk assessment (Table 3). The unbound ∑λ/kdeg does not exceed 0.25, which is the EMA cutoff. Overall, with use of the conservative λ/kdeg cutoff of 0.1 and total Cmax value, the DDI risk after OMP administration was identified without additional consideration of the metabolites.
For reversible CYP2C19 inhibition, the contribution of the metabolites was predicted to be up to 47% of the total interaction risk (Fig. 5; Supplemental Fig. S4). However, the inhibition of CYP2C19 after OMP administration was determined to be mainly attributable to the time dependent inhibition of CYP2C19 by OMP and/or its metabolites. Therefore, the metabolites were predicted to contribute up to 33% of CYP2C19 inhibition, based on unbound concentrations. In contrast, with CYP3A4, based on unbound concentrations, metabolites contribute 88% of the total hepatic CYP3A4 inhibition risk when reversible inhibition is considered, and up to 63% when time dependent inhibition is considered (Fig. 5; Table 3). The percent contribution of metabolites based on their total concentrations are shown in Supplemental Fig. S4. These risk assessments do not consider intestinal CYP3A4 inhibition by systemic metabolites, because it is unclear how intestinal inhibition by systemically formed metabolites should be assessed in DDI risk analysis and whether circulating metabolite concentrations can be used to predict intestinal inhibition of P450s. Regardless, the contribution of metabolites was different for the two P450s inhibited. In addition, the minor metabolite of the quantified metabolites, DM-OMP, was the most relevant in DDI risk assessment.

Predicted relative contribution of OMP and its metabolites to reversible (I/IC50) and irreversible (λ/kdeg) CYP2C19 and CYP3A4 inhibition. The inhibition risk was predicted using unbound Cmax values.
Discussion
Testing of P450 inhibition by metabolites is recommended by the FDA and EMA. This may identify inhibition potential not possessed by the parent drug and aid in predictions of in vivo DDI magnitude and in modeling the time course of the DDI. However, only a few studies have been conducted to evaluate the relative importance and modeling of metabolites in in vivo DDIs (Reese et al., 2008; Templeton et al., 2008; Zhang et al., 2009b; Rowland-Yeo et al., 2010; VandenBrink and Isoherranen, 2010; Yeung et al., 2011). It is unclear whether the relative exposures of metabolites generally correlate with their importance in DDIs. For example, with the CYP3A4 inhibitor itraconazole, the minor metabolite N-desalkyl-itraconazole is predicted to have similar importance in in vivo DDIs as the major metabolite hydroxy-itraconazole (Templeton et al., 2008). Similarly, with the CYP2D6 inhibitor bupropion, erythrobupropion is predicted to have similar role in in vivo DDIs as hydroxybupropion despite its 10-fold lower circulating concentrations (Yeung et al., 2011). To partially address this apparent discrepancy between metabolite exposure and importance in DDIs, consideration of the logP of metabolites was recently recommended for decision trees regarding testing of specific metabolites for inhibition (Yu and Tweedie, 2013). In addition, it was suggested that, for drugs that have structural alerts for TDI, such as alkyamines and epoxides, metabolites should be tested regardless of their exposure (Yu and Tweedie, 2013). However, many compounds, such as omeprazole, are TDIs but do not have obvious structural alerts to trigger DDI evaluation. The aim of this study was to determine, using OMP as an example, whether circulating total or unbound metabolite concentrations could be used to guide in vitro metabolite testing strategy and whether metabolite contribution will aid in predicting in vivo DDIs.
In this study, circulating metabolites of OMP were characterized. As a conservative approach, quantified metabolites were evaluated in vitro if they met any of the criteria for circulating metabolites described by the FDA or EMA. However, DM-OMP only meets the criteria of >25% of the parent using unbound concentrations, and it is <10% of total drug-related material. As such, it is unclear whether testing of this quantitatively minor metabolite would be considered to be necessary. Furthermore, the EMA guidance may not require testing of any of these metabolites. Overall, the analysis of in vivo AUCm/AUCp ratios determined for OMP and its metabolites demonstrates the discrepancies and ambiguity of decision making for metabolite testing. When metabolite evaluation is based solely on metabolite exposure, DM-OMP is the metabolite that would likely be omitted. However, this was the only metabolite that is expected to contribute to in vivo DDIs with CYP2C19 and CYP3A4, and evaluation of OH-OMP, OMP-S, and C-OMP could be considered to be unnecessary. Therefore, it is important to further develop cutoffs for metabolite testing and ensure that the metabolites that are important in DDIs are tested.
Prioritization of metabolite testing based on the lipophilicity of the metabolites has been suggested so that metabolites that are less lipophilic than the parent drug should circulate at concentrations >100% of the parent to warrant testing for reversible inhibition (Yu and Tweedie, 2013). In the present study, all of the metabolites were estimated to be less lipophilic than OMP, except for OMP-S. As such, if metabolite lipophilicity is considered, OMP-S would warrant attention. However, its contribution to the overall DDI risk was predicted to be minimal (5%–8% when unbound Cmax is used); thus, testing of OMP-S could be considered to be unnecessary. With consideration of lipophilicity, DM-OMP would not be studied unless it was flagged for TDI.
Identification of OMP as a TDI is the key element in evaluating DDI risk. OMP is a well-characterized in vivo CYP2C19 inhibitor. It increases the AUC of moclobemide, a CYP2C19 probe, by 120% (Yu et al., 2001), the AUC of proguanil by 50% (Funck-Brentano et al., 1997), and the AUC of diazepam by 25% (Ishizaki et al., 1995) and decreases the formation of the active metabolite of clopidogrel (Angiolillo et al., 2011). OMP also appears to inhibit CYP3A4 in vivo based on a 25% increase in nifedipine AUC (Soons et al., 1992) and 90% increase in carbamazepine AUC (Dixit et al., 2001), but studies with sensitive CYP3A4 probes have not been conducted. The results of this study suggest that better characterization of OMP as a CYP3A4 inhibitor is warranted. Consideration of reversible inhibition alone would not trigger CYP2C19 DDI studies unless all metabolites were considered together with OMP using total Cmax values. However, when OMP was recognized as a TDI of CYP2C19, the predicted in vivo λ/kdeg values were >0.1 using total and >0.25 using unbound Cmax values. Inclusion of the metabolites increased the estimated DDI risk only slightly, and the metabolites were predicted to contribute 20%–50% of the estimated CYP2C19 DDIs. This may be important if more sophisticated prediction methods, such as physiologically based pharmacokinetic (PBPK) models, are used. With CYP3A4, use of total OMP Cmax values for the worst-case scenario risk assessment may provide a sufficient safety margin to ignore metabolite contribution during risk assessment as long as TDI is considered. Total Cmax values usually overpredict in vivo DDIs, and unbound Cmax values are more predictive of in vivo interactions (Obach et al., 2007; Fujioka et al., 2012). Thus, with OMP, the conservative approach of using total Cmax values would likely provide adequate data to trigger in vivo studies without consideration of metabolites.
To our knowledge, time dependent inhibition of CYP3A4 by OMP and the inactivation kinetics of CYP2C19 and CYP3A4 by OMP metabolites have not been previously characterized. The inactivation kinetics of CYP2C19 by OMP has been reported, and the DDI between clopidogrel and OMP has been predicted using OMP data alone (Ogilvie et al., 2011; Boulenc et al., 2012). In addition, OMP-S was previously shown to cause an IC50 shift in vitro with CYP2C19, and the inactivation of CYP2C19 by OMP was proposed to involve DM-OMP (Ogilvie et al., 2011). Our studies demonstrate that DM-OMP is a TDI of CYP2C19, and it is possible that the proposed mechanisms in which a reactive quinoneimine is formed after 5′-O-demethylation by CYP2C19 are responsible for time dependent CYP2C19 inhibition (Ogilvie et al., 2011). However, if quinoneimine formation from DM-OMP is required for CYP2C19 inactivation, it is surprising that the kinact value was the highest for OMP, in comparison with OMP-S and DM-OMP. It is unclear how inactivation proceeds from OMP-S, which has not been shown to undergo O-demethylation. Further studies are required to determine the mechanism of CYP2C19 inactivation by OMP. The reactive quinoneimine formation by CYP3A4 may be responsible for CYP3A4 TDI, because the inactivation rate was faster from DM-OMP than from OMP, and OMP-S did not result in CYP3A4 TDI. Although the DM-OMP formation is generally believed to be by CYP2C19 (Andersson and Weidolf, 2008), the data in CYP3A4 supersomes show that CYP3A4 TDI does not require CYP2C19. This suggests that either CYP3A4 forms sufficient quantities of DM-OMP that is not released from CYP3A4 active site and results in CYP3A4 TDI or that the quinoneimine is not responsible for CYP3A4 inhibition. The qualitative differences between CYP2C19 and CYP3A4 inactivation by OMP and its metabolites are of interest, because they demonstrate that P450 inactivation mechanisms and relative metabolite contributions cannot be easily generalized and extrapolated. The data also show that metabolites formed by one P450 (DM-OMP by CYP2C19 and OMP-S by CYP3A4) may be relevant in inhibition of other P450s.
OMP has a short half-life and its metabolites, with the exception of OMP-S, follow formation rate–limited kinetics. Because of its short half-life, use of time-varying DDI models may be beneficial for quantitative DDI predictions of OMP. The risk predictions provided here should not be directly used for quantitative DDI predictions. However, the metabolite to parent concentration ratio does not change as a function of time for OH-OMP, DM-OMP, and C-OMP; thus, the Cmax ratios should provide a reasonable assessment of the relative importance of these metabolites in CYP2C19 and CYP3A4 inhibition. These predictions were made on the basis of plasma concentrations in CYP2C19-extensive metabolizers after a 20-mg dose of OMP in a four-drug cocktail. Because OMP inhibits CYP2C19, the metabolite ratios may change with increasing doses and multiple administrations, and better characterization of metabolite disposition after different treatment regimens is required for PBPK models of OMP. In addition, because OH-OMP, DM-OMP, and C-OMP are also reversible inhibitors of CYP2C19 and CYP3A4 and their [I]/Ki ratios were, in many cases, higher than those for OMP, it may be necessary to account for inhibitor-inhibitor interactions between the metabolites and OMP in quantitative DDI predictions. Further work is required to develop the kinetic theory for multiple inactivators according to the theory provided for alkylamines (Zhang et al., 2009a).
In conclusion, the results of this study show that identification of TDIs during drug development is critically important for DDI risk assessment. On the basis of the obtained data, DM-OMP is responsible for the majority of hepatic CYP3A4 inhibition, whereas metabolites are responsible for <50% of the overall CYP2C19 inhibition. Although metabolites may contribute to in vivo DDIs, their importance may not be related to their relative abundance in plasma. As such, better models need to be developed to prioritize metabolite testing in DDI assessment.
Authorship Contributions
Participated in research design: Shirasaka, Sager, Isoherranen, Lutz.
Conducted experiments: Shirasaka, Sager, Lutz.
Performed data analysis: Shirasaka, Sager, Lutz, Davis.
Wrote or contributed to the writing of the manuscript: Shirasaka, Sager, Lutz, Isoherranen.
Footnotes
- Received February 23, 2013.
- Accepted April 25, 2013.
Y.S. and J.E.S. contributed equally to this article.
This work was supported by the National Institutes of Health [Grants P01 GM32165 (to J.D.L., CD., N.I.) and T32 GM007750 (to J.E.S. and University of Washington Clinical Research Center)]; the National Institutes of Health National Center for Advancing Translational Sciences [Grant UL1TR000423]; the Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad [H23-694 (to Y.S.)]; and a Grant-in-Aid for Scientific Research [21790147 (to Y.S.)].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the plasma concentration–time curve
- Cmax
- maximum concentration in plasma
- C-OMP
- carboxyomeprazole
- DDI
- drug-drug interaction
- DM-OMP
- 5′-O-desmethylomeprazole
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- fu,p
- fraction of drug unbound in plasma
- fu,mic
- fraction of drug unbound in microsomes
- OH-OMP
- 5-hydroxyomeprazole
- HLM
- human liver microsome
- [I]
- inhibitor concentration
- IC50
- inhibitor concentration that results in half-maximal inhibition
- kdeg
- rate constant for in vivo P450 degradation
- Ki
- inhibition constant
- KI
- inhibitor concentration when the rate of inactivation reaches half of kinact
- Km
- Michaelis constant
- kinact
- maximum time-dependent inactivation rate
- λ
- apparent inactivation rate at a given inhibitor concentration
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- OMP
- omeprazole
- OMP-S
- omeprazole sulfone
- P450
- cytochrome P450
- TDI
- time dependent inhibition or inhibitor
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}