


Visual Overview

Abstract
Although intestinal metabolism plays an important role in drug disposition, early predictions of human outcomes are challenging, in part because of limitations of available in vitro models. To address this, we have evaluated three in vitro models of human intestine (microsomes, permeabilized enterocytes, and cryopreserved intestinal mucosal epithelium) as tools to assess intestinal metabolism and estimate the fraction escaping gut metabolism (fg) in drug discovery. The models were tested with a chemically diverse set of 32 compounds, including substrates for oxidoreductive, hydrolytic, and conjugative enzymes. Liquid chromatography–high-resolution mass spectrometry was used to quantify substrate disappearance [intrinsic clearance (CLint)] and qualify metabolite formation (quantitative-qualitative bioanalysis). Fraction unbound in the incubation (fu,inc) was determined by rapid equilibrium dialysis. Measured in vitro results (CLint and fu,inc) were supplemented with literature data [passive Caco-2 apical to basolateral permeability, enterocyte blood flow, and intestinal surface area (A)] and combined using a midazolam-calibrated Qgut model to predict human fg values. All three models showed reliable CYP and UDP-glucuronosyltransferase activities, but enterocytes and mucosa may offer advantages for low-clearance compounds and alternative pathways (e.g., sulfation, hydrolases, and flavin-containing monooxigenases). Early predictions of human fg values were acceptable for the high-fg compounds (arbitrarily fg > 0.7). However, predictions of low- and moderate-fg values (arbitrarily fg < 0.7) remain challenging, indicating that further evaluation is needed (e.g., saturation effects and impact of transporters) but not immediate compound avoidance. Results suggest that tested models offer an additional value in drug discovery, especially for drug design and chemotype evaluation.
Significance Statement We found that cellular models of the human gut (permeabilized enterocytes and cryopreserved intestinal mucosa) offer an alternative to and potential advantage over intestinal microsomes in studies of drug metabolism, particularly for low-clearance compounds and alternative pathways (e.g., sulfation, hydrolases, and flavin-containing monooxigenases). The predictivity of human fraction escaping gut metabolism for common CYP and UDP-glucuronosyltransferase substrates based on the Qgut model is still limited, however, and appropriate further evaluation is recommended.
Introduction
Metabolism of the intestinal wall plays an important role in the presystemic disposition of oral drugs, including first-pass elimination (Hall et al., 1999; Jones et al., 2016; Peters et al., 2016; Yau et al., 2017), drug interactions (Tachibana et al., 2010; Alqahtani et al., 2018; Yamada et al., 2020), and bioactivation (Dalvie et al., 2008; Yang et al., 2019). Early evaluation of the fraction escaping gut metabolism (fg), a significant contributor to oral bioavailability (F) together with fraction absorbed (fa) and fraction escaping liver metabolism (fh) (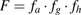 ), is particularly relevant in drug discovery settings, both for predictions of human efficacious dose (Gertz et al., 2010; Peters et al., 2016) and evaluation of prodrugs (Williams et al., 2011; Nishimuta et al., 2014; Takahashi et al., 2019). Preferably, human intestinal drug metabolism and corresponding fg values could be evaluated in vitro, reducing the unnecessary animal testing [3R; EMA/CHMP/CVMP/JEG-3Rs/450091/2012; (Burden et al., 2015)] and accelerating drug discovery.
), is particularly relevant in drug discovery settings, both for predictions of human efficacious dose (Gertz et al., 2010; Peters et al., 2016) and evaluation of prodrugs (Williams et al., 2011; Nishimuta et al., 2014; Takahashi et al., 2019). Preferably, human intestinal drug metabolism and corresponding fg values could be evaluated in vitro, reducing the unnecessary animal testing [3R; EMA/CHMP/CVMP/JEG-3Rs/450091/2012; (Burden et al., 2015)] and accelerating drug discovery.
Despite the need for an early and accurate evaluation of intestinal drug metabolism, common in vitro models, such as intestinal microsomes and tissue slices, present drug discovery scientists with a difficult choice between a “too simple” and “too complex” model (Fig. 1). Intestinal microsomes generally capture CYP (Gertz et al., 2010) and UDP-glucuronosyltransferase (UGT) activities (Cubitt et al., 2009; Kosaka et al., 2011; Gill et al., 2012) but lack many cytosolic [e.g., sulfotransferases, aldehyde oxidase (AO), and various hydrolyses], mitochondrial (e.g., monoamineoxidase), and often even soluble endoplasmic enzymes [carboxylesterases (CESs) (Wang et al., 2018)]. Moreover, a complex process of gut tissue procurement and subcellular fractionation may result in enzyme inactivation (Mohri and Uesawa, 2001; Hatley et al., 2017a,b), leading to underprediction of metabolic clearance and batch-to-batch variability. In contrast to microsomes, intestinal precision-cut tissue slices offer preserved tissue architecture and a complete set of drug-metabolizing enzymes (van de Kerkhof et al., 2006, 2008; de Graaf et al., 2010). Recent advances in organotypic three-dimensional cell cultures—for example, intestinal organoids (Lu et al., 2017; Almeqdadi et al., 2019) and gut-on-a-chip systems (Guo et al., 2018)—are equally promising. However, the enduring challenges of ethical procurement of fresh human tissue (Allen et al., 2010), tissue slicing and culturing techniques (Li et al., 2016), and interindividual variability (Quignot et al., 2019) render these models better suited for in-depth mechanistic studies with a single compound rather than chemotype evaluations performed in drug discovery.

An overview of available in vitro models of the human intestine.
We hypothesized that emerging cellular models of the human gut like primary enterocytes (Ho et al., 2017; Wong et al., 2018) and intestinal mucosal epithelium (Li et al., 2018a) may bridge the gap and offer an in vitro model of “intermediate complexity” suitable for drug discovery applications (Fig. 1) similar to cryopreserved hepatocytes as representative model of human liver (Hengstler et al., 2000; Li, 2007). Although they can potentially capture relevant biology of drug-metabolizing enzymes in the gut wall, these models offer diminished interindividual variability through donor pooling and can be used in automated screening assays (Jacobson et al., 2007). To test this hypothesis, we have assayed 32 test compounds (with versatile biotransformation pathways and a range of human observed fg values) in human intestinal microsomes, permeabilized and co-factor supplemented MetMax human enterocytes (Li et al., 2018b), and cryopreserved human intestinal mucosal epithelium (Li et al., 2018a). Permeabilized human enterocytes (MetMax) were selected based on reported higher activities of several drug-metabolizing enzymes compared with corresponding conventional (nonpermeabilized) enterocytes (Li et al., 2018b) and a relatively simple biochemical assay format that resembles microsomal incubations (Li et al., 2018b). Cryopreserved intestinal mucosa was included in our evaluation because of a presence of several key intestinal cell types (Li et al., 2018a), including enterocytes, goblet cells, enteroendocrine paneth, and microfold cells, potentially broadening the scope of available drug-metabolizing enzymes and approaching level of complexity only available in organotypic gut models (Maresca et al., 2018). As a differentiation compared with intestinal mucosa, permeabilized enterocytes offer an increased cellular permeability and biochemical assay environment (i.e., no need for cell counting and culturing) (Li et al., 2018a,c), possibly enabling drug metabolism studies with poorly permeable and cytotoxic compounds (e.g., cancer chemotherapeutics).
To enable simultaneous insight into both quantitative disappearance of the parent drug and qualitative appearance of drug metabolites (QuanQual), all study samples were analyzed with liquid chromatography (LC)–high-resolution mass spectrometry (HRMS) analytical methods. In addition to evaluation of intrinsic clearance (CLint) and biotransformation pathways, a midazolam-calibrated Qgut model (Yang et al., 2007; Gertz et al., 2010) was used for early predictions of human fg values. Similarities and differences between available in vitro models are discussed, enabling an informed approach to intestinal metabolism evaluation in drug discovery settings.
Materials and Methods
Reagents and Chemicals
Uridine 5′-diphosphoglucuronic acid trisodium salt (UDPGA), NADPH, alamethicin from Trichoderma viride, adenosine 3-phosphate 5-phosphosulfate, diclofenac sodium salt, midazolam hydrochloride, felodipine, S-(+)-mephenytoin, raloxifene hydrochloride, lovastatin, nisoldipine, nifedipine, methadone, quinidine, testosterone, benzydamine hydrochloride, cisapride, cyclosporin, terbutaline, sildenafil, verapamil, trazodone, atorvastatin, simvastatin, buspirone, rifabutin, saquinavir, terfenadine, zolpidem, repaglinide, indinavir, alprazolam, carbazeran, enalapril, ramipril, and dabigatran etexilate were purchased from Sigma-Aldrich (St. Louis, MO). NADPH-regenerating system containing NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase was purchased from Corning (Wiesbaden, Germany). Organic solvents were of LC–mass spectrometry (MS) or higher quality grade and were acquired from VWR International (Radnor, PA) or Fisher Scientific UK Ltd (Loughborough, UK). Purified water was obtained from Milli-Q Integral 5 Water Purification System (Merck KGaA, Darmstadt, Germany). Phosphate buffer, HQM Hepatocyte/Enterocyte Incubation Medium, and Co-Factor N for MetMax were purchased from In Vitro ADMET Laboratories (Columbia, MD).
In Vitro Models
Human intestinal microsomes (lot GYC; mixed-sex, n = 6; from upper third of the intestine, mostly duodenum) were purchased from BioreclamationIVT (Baltimore, MD) and stored at −80°C until use. Permeabilized human enterocytes (I PHEX MetMax-B, High Performance Pooled Human Enterocytes Phase I/II, lot PHEX3107; mixed-sex, n = 10; prepared from the duodenum, jejunum, and ileum; stored in −80°C) and cryopreserved human intestinal mucosal epithelium (PCHIM pooled; lot PCHIM6031; mixed-sex, n = 5; prepared from the jejunum; stored in liquid nitrogen) were supplied by In Vitro ADMET Laboratories. Certificates of Analysis stated that CYP3A4-mediated rates of testosterone hydroxylation (formation of 6β-hydroxytestosterone at 200 µM of the substrate) were 163, 367, and 612 pmol/min per milligram or 106 cells in intestinal microsomes, permeabilized enterocytes, and cryopreserved mucosa, respectively. Reported rates of 7-hydroxycoumarine glucuronidation were 147, 81, and 62 pmol/min per milligram and 106 cells in intestinal microsomes, permeabilized enterocytes, and cryopreserved mucosa, respectively. At the time when functional experiments were performed, knowledge of enzyme and transporter protein expression in these models was unknown.
In Vitro Assays
Incubations with In Vitro Models of the Human Gut Wall.
Incubations with human intestinal microsomes were performed with 1 mg/ml of microsomal protein and NADPH-regenerating system (containing 5.5 mM MgCl2) in 100 mM phosphate buffer (pH 7.4). For compounds that predominantly undergo glucuronidation, microsomes were preincubated on ice water for 20 minutes with a pore-forming peptide, alamethicin (at a concentration equivalent to 5% microsomal protein) (Fisher et al., 2000), and incubations were supplemented with UDPGA (2.5 mM). Optimization tests showed that preincubation of microsomes with alamethicin, which is beneficial for optimal UGT activity (Walsky et al., 2012), slightly diminished CYP activities, leading to variability of low-clearance CYP substrates. Therefore, the inclusion of alamethicin and UDPGA was dependent on the compound used and its predominant biotransformation pathway. UDPGA was included for experiments involving testosterone, diclofenac, and raloxifene. Incubations with permeabilized human enterocytes were performed with 500,000 cells/ml and Co-Factor N for MetMax (a mixture of enzyme cofactors) in HQM Hepatocyte/Enterocyte Incubation Medium (Li et al., 2018b). Incubations with intestinal mucosa were performed with 1 mg/ml total protein (determined by manufacturer) in HQM Hepatocyte/Enterocyte Incubation Medium (Li et al., 2018a). Incubations contained a maximum of 0.5% acetonitrile and 0.1% of DMSO.
All incubations were carried out at 37°C with continuous shaking at 700 rpm (Eppendorf ThermoMixer C; Thermo Fisher Scientific, Bremen, Germany). Determination of CLint and analysis of biotransformation pathways were performed at 1 μM final substrate concentration, and incubation time was 1–60 minutes (six time points). Saturation and enzyme kinetic experiments with indinavir and raloxifene were performed at 0.1–100 μM (seven to eight concentrations) for 10 minutes. Typically, incubations were performed in a 96–deep well plates with temporal sampling and an incubation volume of 50 μl per individual time point. Reactions were prewarmed in an incubator for 5 minutes and initiated with the addition of test substrate. After the required time, reactions were terminated by mixing one volume of incubation mixture with two volumes of ice-cold acetonitrile (containing internal standard) and one volume of ice-cold water. Terminated reactions were placed in a fridge for 20 minutes and then centrifuged for 10 minutes at 4°C and 3000 rpm. Aliquots of the supernatants were separated from the pellets using a Microlab STAR liquid handling system (Hamilton Robotics Inc., NV) and analyzed by LC-HRMS. All incubations were performed in at least two replications (n = 2−6), with midazolam as a positive control.
Determination of Fraction Unbound in the Incubation.
The rapid equilibrium dialysis (RED) method (ThermoFisher Scientific, Rockford, IL) was used to determine the degree of nonspecific binding of the compounds to intestinal models during in vitro incubations. Incubations were carried out according to manufacturer’s instructions at 37°C for 4 hours with 10 μM of the test compound (compound concentration was increased compared with functional assays to enable bioanalysis of highly bound compound). Protein or cell concentrations of intestinal models were the same as in the functional assays. Prior to fraction unbound in the incubation (fu,inc) determinations, enzymes were inactivated by 48-hour preincubation at 5°C, and enzyme inactivity was confirmed by the lack of midazolam metabolite formation after 4 hours of incubation. For highly bound compound, readers are also advised to evaluate longer equilibration times and/or alternative methods (Chen et al., 2019).
Analytical Methods
Samples were analyzed by LC-HRMS to monitor for both disappearance of test compound and appearance of drug metabolites (QuanQual approach). LC-HRMS system consisted of PAL autosampler (set to 8°C; CTC Analytics AG, Zwingen, Switzerland), Accela 1250 liquid chromatograph (Thermo Fisher Scientific), MistraSwitch thermostated column compartment (set to 50°C; MayLab Analytical Instruments GmbH, Germany), and Q Exactive orbitrap mass spectrometer (Thermo Fisher Scientific). Two analytical methods were used: 1) a 5-minute LC gradient with HRMS acquisition for QuanQual analysis using the Phenomex Kinetex C18 column (50 × 2.1 mm, 5 µm) (Phenomenex, Inc., Macclesfield, United Kingdom) and 2) a 15-minute LC gradient for metabolite identification with HRMS and data-dependent MS/MS acquisition using a Phenomenex Kinetex C18 column (100 × 3.0 mm, 2.6 µm). Both methods used either 0.1% formic acid in water (low pH conditions) or 10 mM ammonium bicarbonate, pH 8.5 (high pH conditions), as eluent A and 0.1% formic acid in acetonitrile as eluent B. The LC gradient for the 5-minute method was 0–0.4 minutes, 5% B; 0.4–2.5 minutes, 5% → 95% B; 2.5–4.5 minutes, 95% B; 4.5–4.6 minutes, 95→5% B; and 4.6–5.6 minutes, 5% B. The LC gradient for the 15-minute method was 0–0.8 minutes, 5% B; 0.8–12.0 minutes, 5→95% B; 12.0–13.0 minutes, 95% B; 13.0–13.1 minutes, 95→5% B; and 13.1–15.0 minutes, 5% B. The flow rate of the mobile phase was 0.6 ml/min for the 5-minute method and 0.5 ml/min for the 15-minute method with an injection volume of 10 μl.
Data Analysis
Chemical structures and biotransformation schemes were prepared with ACD/ChemSketch 2018.1 (ACD Laboratories). Graphs, linear and nonlinear regression, and statistical analysis were performed with GraphPad Prism 8.1 (GraphPad Software).
Determination of Intestinal CLint.
CLint (microliters per minute per milligram of total protein or microliters per minute per 106 cells) was determined by following the depletion of test compound over incubation time. Compound elimination rate constant (kel; 1/min) was determined by using nonlinear regression and monoexponential decay equation (GraphPad Prism 8.1; GraphPad Software, San Diego CA). CLint was calculated as (eq. 1): In which m(enzyme source) represents either total amount of protein (microsomal and mucosal incubations) or total number of cells (enterocyte incubations) used in the assay, and Vinc represents the volume of incubation (microliters). Estimated lower limit of quantification for CLint determination was 1 μl/min per 106 cells or 1 μl/min per milligram protein, corresponding to 97% of test compound remaining after a 60-minute incubation. Results are presented as mean ± S.E.
In which m(enzyme source) represents either total amount of protein (microsomal and mucosal incubations) or total number of cells (enterocyte incubations) used in the assay, and Vinc represents the volume of incubation (microliters). Estimated lower limit of quantification for CLint determination was 1 μl/min per 106 cells or 1 μl/min per milligram protein, corresponding to 97% of test compound remaining after a 60-minute incubation. Results are presented as mean ± S.E.
Analysis of Intestinal Biotransformation Pathways.
For drug metabolite profiling, acquired LC-HRMS raw data were processed by Compound Discoverer 3.1 (Thermo Fisher Scientific) using a targeted workflow. Theoretical drug metabolites were predicted from various oxidoreductive (oxidation, desaturation, hydration, hydrolysis, dealkylation, etc.) and conjugative (glucuronidation, sulfation, glutathione conjugation, etc.) biotransformation pathways, including all combinations up to three steps, and generated mass ions were searched against experimentally obtained data. Metabolite identity was established by a combination of mass tolerance (<10 ppm), absence in blank incubations, kinetics of metabolite formation, matching isotope patterns, and, when appropriate, analysis of MS/MS data. Because the parent compound and its metabolites may show different ionization responses in mass spectrometer, obtained metabolite data are qualitative or semiquantitative at best (Blanz et al., 2017).
Prediction of Human fg Values Based on In Vitro Data.
Human fg values were predicted based on midazolam-calibrated Qgut model (Rostami-Hodjegan and Tucker, 2002; Yang et al., 2007; Gertz et al., 2010). In addition to CLint values experimentally measured in this work, Qgut model requires measurements of intestinal permeability [either apparent permeability (Papp; nanometers per second) or effective permeability (Peff; micrometers per second)], intestinal surface area (Aint; squared meter), and enterocytic blood flow (Qent; liters per hour). Because measured in vivo Peff values are rarely available in drug discovery settings, and conversion between Papp and Peff could introduce additional errors (Gertz et al., 2010), we have directly used literature in vitro Papp values (nanometers per second) from Caco-2 A→B passive permeability assays (Wu et al., 2000; Polli et al., 2001; Hochman et al., 2004; Gertz et al., 2010; Lee et al., 2017). Literature Caco-2 Papp values were supplemented with experimentally measured values for ramipril, enalapril, terbutaline, terfenadine, raloxifene, benzydamine, and carbazeran (Cyprotex, Alderley Park, UK). Because dabigatran etexilate is known to undergo esterase-mediated hydrolysis in the Caco-2 cells (Ishiguro et al., 2014), we have used the passive Papp value obtained in the optimized assay, which was performed in the presence of both CES inhibitor [bis(p-nitrophenyl) phosphate)] and P-gp inhibitor (cyclosporin A) (Ishiguro et al., 2014). Average values of 200 m2 for Aint (Gertz et al., 2010) and 18 l/h for Qent were used (Granger et al., 1980).
Permeability clearance (CLperm), a hybrid parameter of Papp and Aint, was calculated as (eq. 2):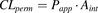 CLperm and Qent were combined to form a Qgut, a hybrid parameter of permeability through the enterocyte membrane and Qent (eq. 3):
CLperm and Qent were combined to form a Qgut, a hybrid parameter of permeability through the enterocyte membrane and Qent (eq. 3):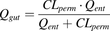 The fg is then calculated by the following equation (eq. 4):
The fg is then calculated by the following equation (eq. 4): In which fu,gut and fu,inc represent the fraction unbound in the gut and enterocyte incubations, respectively. The errors of CLint measurements were propagated into calculated fg values by using the following equation (eq. 5) (Taylor, 1996):
In which fu,gut and fu,inc represent the fraction unbound in the gut and enterocyte incubations, respectively. The errors of CLint measurements were propagated into calculated fg values by using the following equation (eq. 5) (Taylor, 1996):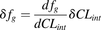
In which δfg and δCLint are errors of fg and CLint, respectively, and dfg/dCLint is the first derivative of the fg function over CLint.
To enable direct comparison of fg predictivity between three intestinal models (intestinal microsomes, permeabilized enterocytes, and mucosa) and account for batch-to-batch variations, calculated fg values were calibrated to in vivo observed fg value of midazolam [0.55 ± 0.04; mean ± S.E.; n = 6; (Paine, 1996; Galetin et al., 2006; Gertz et al., 2008, 2010; Nishimuta et al., 2011; Karlsson et al., 2013)]. Midazolam calibration resulted in microsomal-, enterocytic-, and mucosal-predicted fg values to be corrected +9.1%, +18.2%, and −12.7%, respectively. Minimal and maximal predicted fg values were arbitrarily set to 0.01 and 1, respectively.
Evaluation of Observed Human fg Values.
Observed in vivo human fg values were obtained from the literature for alprazolam, atorvastatin, buspirone, cisapride, cyclosporine, felodipine, indinavir, lovastatin, methadone, midazolam, nifedipine, nisoldipine, quinidine, raloxifene, repaglinide, rifabutin, saquinavir, sildenafil, simvastatin, tacrolimus, terfenadine, trazodone, triazolam, verapamil, and zolpidem (Paine et al., 1996; Galetin et al., 2006; Yang et al., 2007; Gertz et al., 2008, 2010; Mizuma, 2009; Nishimuta et al., 2011; Karlsson et al., 2013) (Table 1). The majority of human fg values was estimated through interpretation of clinical PK results (e.g., intravenous and oral drug administration) and corresponding in vitro data. A smaller subset of observed human fg values, including CYP3A4 substrates midazolam, felodipine, buspirone, quinidine, and lovastatin, were obtained via the “grapefruit juice method” (Gertz et al., 2008; Hanley et al., 2011). When multiple estimates of observed fg values were published, the arithmetic mean ± S.D. was used for comparisons with predicted fg values. Because clinical PK results may be influenced by a number of variables, including subject number, age, sex, ethnicity, drug dose, disease state, and polymorphisms of enzymes and transporters, readers should note that reported human fg values are estimates at the current level of knowledge. Although more sophisticated meta-analyses of clinical data, including interstudy and interindividual variability and weighted means, were performed previously (Gertz et al., 2008), this was beyond the scope of current work. If literature values were not available, observed fg values were estimated from available clinical PK data and complementary in vitro data using the following equation (eq. 6):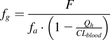 In which F is the absolute bioavailability of the drug (fraction 0–1), fa is the fraction of the dose absorbed (fraction 0–1), Qh is the human liver blood flow [1450 ml/min for a 70-kg individual (Davies and Morris, 1993)], and CLblood is the systemic clearance of the drug (assumption CLblood ≈ CLliver). As an example, clinical PK parameters for diclofenac were taken from (Willis et al., 1979; Hinz et al., 2005), and fa was assumed to be 1 because of high Caco-2 Papp value (>100 nm/s). Similar evaluations were performed for enalapril (Davies et al., 1984), raloxifene (Mizuma, 2009), testosterone (Tauber et al., 1986), dabigatran etexilate (Blech et al., 2008; Ishiguro et al., 2014), and benzydamine (Baldock et al., 1991). Human fg values of carbazeran [F < 5% (Kaye et al., 1984)], S-mephenytoin [F < 5% and large interindividual variability (Wedlund et al., 1985)], and terbutaline could not be estimated based on available data.
In which F is the absolute bioavailability of the drug (fraction 0–1), fa is the fraction of the dose absorbed (fraction 0–1), Qh is the human liver blood flow [1450 ml/min for a 70-kg individual (Davies and Morris, 1993)], and CLblood is the systemic clearance of the drug (assumption CLblood ≈ CLliver). As an example, clinical PK parameters for diclofenac were taken from (Willis et al., 1979; Hinz et al., 2005), and fa was assumed to be 1 because of high Caco-2 Papp value (>100 nm/s). Similar evaluations were performed for enalapril (Davies et al., 1984), raloxifene (Mizuma, 2009), testosterone (Tauber et al., 1986), dabigatran etexilate (Blech et al., 2008; Ishiguro et al., 2014), and benzydamine (Baldock et al., 1991). Human fg values of carbazeran [F < 5% (Kaye et al., 1984)], S-mephenytoin [F < 5% and large interindividual variability (Wedlund et al., 1985)], and terbutaline could not be estimated based on available data.
Test compounds used in this study (n = 32), sorted by the increasing avg. observed human fg value
Compounds were selected based on a wide range of observed fg values, different biotransformation pathways, and physicochemical properties. Maximum dose, aqueous solubility, and logD values were reported from (Benet et al., 2011) and other publicly available sources (Drug Bank, https://www.drugbank.ca/; ChEMBL, https://www.ebi.ac.uk/chembl/).
Comparison of Observed and Predicted Human fg Values.
Observed and predicted human fg values were compared based on geometric mean fold error (GMFE). Furthermore, based on the range of observed human fg values, compounds were arbitrarily classified into low (fg 0–0.3), moderate (fg 0.31–0.70), and high (fg 0.71–1.0) groups. GMFE values, classification success (based on predicted fg values), and prediction bias were calculated for each group.
Saturation and Enzyme Kinetic Experiments.
To assess the effects of enzyme saturation, the apparent CLint for indinavir and raloxifene was measured across the range of substrate concentrations (0.1–100 μM) in human permeabilized enterocytes. The sum of LC-HRMS peak areas for all observed metabolites was used to estimate the apparent Km values. The velocity of the enzymatic reaction (v) was plotted against the substrate concentration ([S]), and data were fitted to either Michaelis-Menten ( ) or substrate inhibition equations (
) or substrate inhibition equations (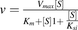 ), in which Km is the Michaelis-Menten constant, Vmax is the limiting reaction velocity, and Ksi is the substrate inhibition constant. Because drug metabolites were not quantified with authentic standards but rather observed with QuanQual bioanalytical approach, Vmax values are qualitative and expressed at the metabolite peak area per minute/106 cells. The Km values were also estimated based on the substrate disappearance data. Observed elimination rate constants (kel, 1/min) were plotted against the substrate concentration ([S], micromolars), and data were fitted to the following equation:
), in which Km is the Michaelis-Menten constant, Vmax is the limiting reaction velocity, and Ksi is the substrate inhibition constant. Because drug metabolites were not quantified with authentic standards but rather observed with QuanQual bioanalytical approach, Vmax values are qualitative and expressed at the metabolite peak area per minute/106 cells. The Km values were also estimated based on the substrate disappearance data. Observed elimination rate constants (kel, 1/min) were plotted against the substrate concentration ([S], micromolars), and data were fitted to the following equation:  , in which kel,max is the theoretical maximal kel at an infinitesimally low concentration of substrate (Obach and Reed-Hagen, 2002).
, in which kel,max is the theoretical maximal kel at an infinitesimally low concentration of substrate (Obach and Reed-Hagen, 2002).
Results
Selection of the Test Compounds.
Our 32 test compounds were selected based on a range of observed human fg values as well as different biotransformation pathways and diverse drug-like properties (Table 1). Most of the previously reported observed human fg values were estimated through integration of clinical PK results (e.g., intravenous and oral drug administration) and corresponding in vitro data (e.g., solubility and permeability). A smaller subset of observed human fg values for midazolam, felodipine, buspirone, quinidine, and lovastatin were obtained via the “grapefruit juice method,” an approach that utilizes CYP3A4 inhibition of grapefruit juice ingredients (Hanley et al., 2011). Although the majority of human fg values were obtained from the literature (see Table 1 for references), values for enalapril, raloxifene, testosterone, dabigatran etexilate, and benzydamine were estimated based on available human PK results and corresponding in vitro data (see Data Analysis for details). The observed fg values for test compounds ranged from 0.05 to 0.98, with median and average values of 0.57 and 0.53, respectively. Whereas most of the test compounds undergo oxidative metabolism (especially by CYP3A4), raloxifene, testosterone, and terbutaline undergo conjugations (glucuronidation and sulfation), and dabigatran etexilate, ramipril, and enalapril are hydrolyzed by hydrolytic enzymes. In addition to CYP-mediated oxidations, benzydamine is predominantly oxidized by FMOs and carbazeran by AO. Regarding the drug-like and physicochemical properties, tested compounds showed a diverse range of values (Table 1) (range; mean ± S.D.): Caco-2 permeability (1.37–722; 167 ± 150 nm/s), logD at pH 7.4 (–1.44–5.05; 2.50 ± 1.65), aqueous solubility (<0.01–213 mg/ml), and maximum dose (2–500 mg).
In Vitro CLint.
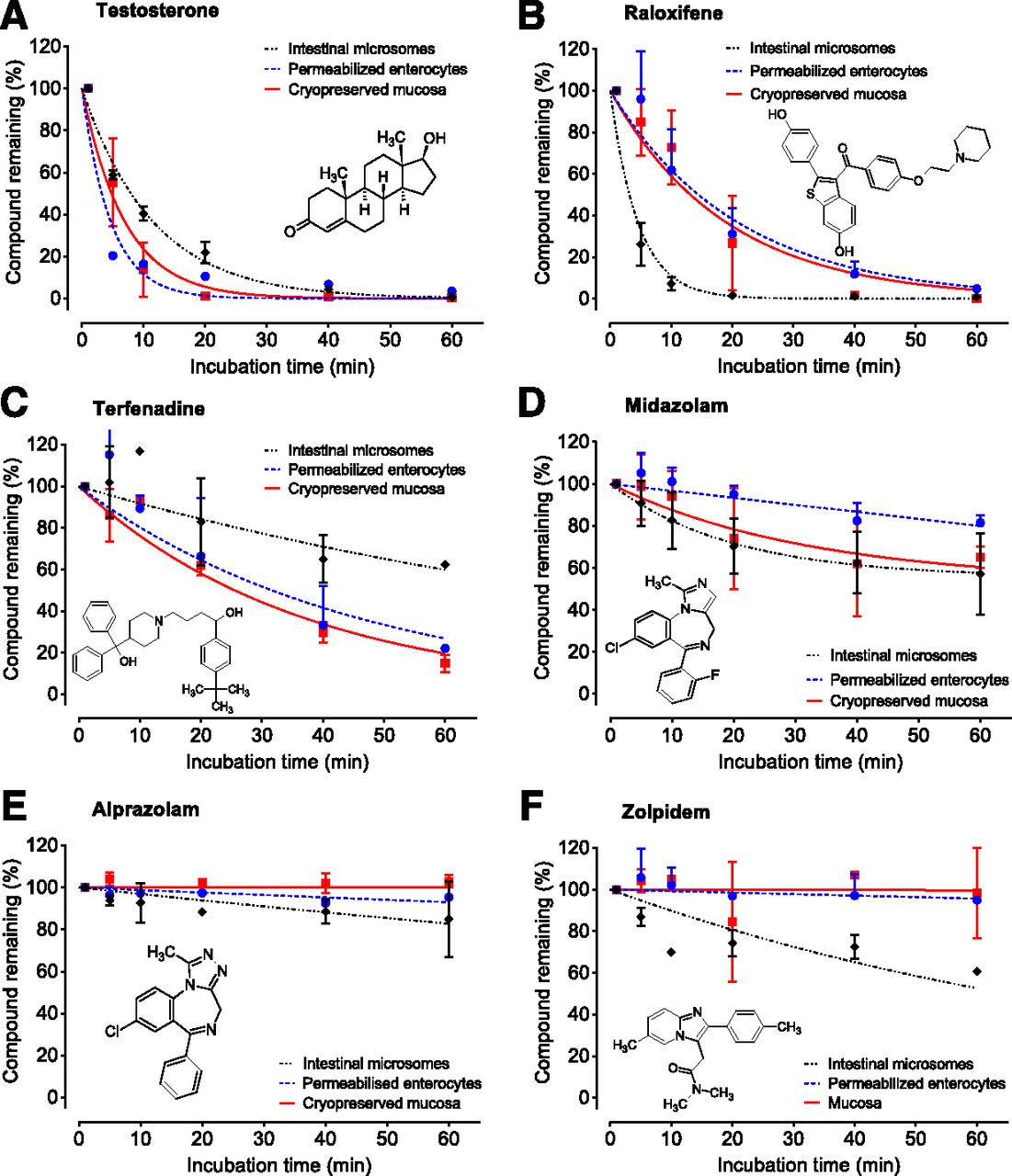
The CLint values were measured for all 32 test compounds in intestinal microsomes, permeabilized enterocytes, and intestinal mucosa. Experiments were performed at 1 μM of substrate and 1–60 minutes of incubation, resembling assay conditions typical for drug discovery. Numerical results are presented in Table 2 and selected graphical examples in Fig. 2 (for all graphs, see Supplemental Figs. 1–6). For test substrates predominantly metabolized by CYPs, the observed rate of metabolism was generally comparable between models, although average values of CLint were 2-fold higher in permeabilized enterocytes and intestinal mucosa (44.74 and 46.46 μl/min per milligram or 106 cells, respectively, compared with 20.85 μl/min per milligram in microsomes). Higher observed CYP metabolism in permeabilized enterocytes and intestinal mucosa is aligned with formation rates of 6β-hydroxytestosterone reported by vendors in the corresponding Certificates of Analysis. However, a few exceptions to this trend were observed, with S-mephenytoin and zolpidem showing higher CLint in intestinal microsomes. Benzydamine, an FMO substrate (Lang and Rettie, 2000), showed a significantly higher CLint in permeabilized enterocytes and mucosa compared with microsomes (Table 2). Carbazeran, a high-turnover substrate for human AO (Kaye et al., 1984), showed very low clearance in all three intestinal models. Test substrates for hydrolytic enzymes, namely dabigatran etexilate, ramipril, and enalapril, showed variable results (Table 2). Although CLint of dabigatran etexilate was 10-fold higher in permeabilized enterocytes and intestinal mucosa, metabolism of ramipril and enalapril was minimal in all three models. Raloxifene and testosterone, test substrates for glucuronidation and sulfation, generally exhibited high CLint in all three models (typically >100 μl/min per milligram or 106 cells), although CLint of testosterone was still 5-fold higher in permeabilized enterocytes and intestinal mucosa. Although 60-minute incubation time enabled a wide dynamic range of the CLint assays, one may consider shorter or longer incubation times depending on the properties of the chemotype, such as, for example, up to 20 or 120 minutes for evaluation of prodrugs (e.g., CLint > 300 µl/min per milligram) or low-clearance compounds (CLint < 3 µl/min per milligram), respectively.
CLint values (microliters per minute per milligram protein or 106 cells) of 32 test compounds using intestinal microsomes, permeabilized enterocytes, and intestinal mucosa and their respective observed in vivo fg value
CLint values were measured in at least three independent experiments, and results are presented as the mean ± S.E.

An example of CLint measurements performed for compounds with low (A and B), medium (C and D), and high (E and F) observed human fg values. Experiments were performed with human intestinal microsomes, permeabilized enterocytes, and cryopreserved mucosa. Presented data points are mean values ± S.D. from at least three independent experiments. Observed human fg values for the given compounds were (order from A to F) 0.05, 0.05, 0.40, 0.55, 0.94, and 0.79, respectively.
Biotransformation Pathways.
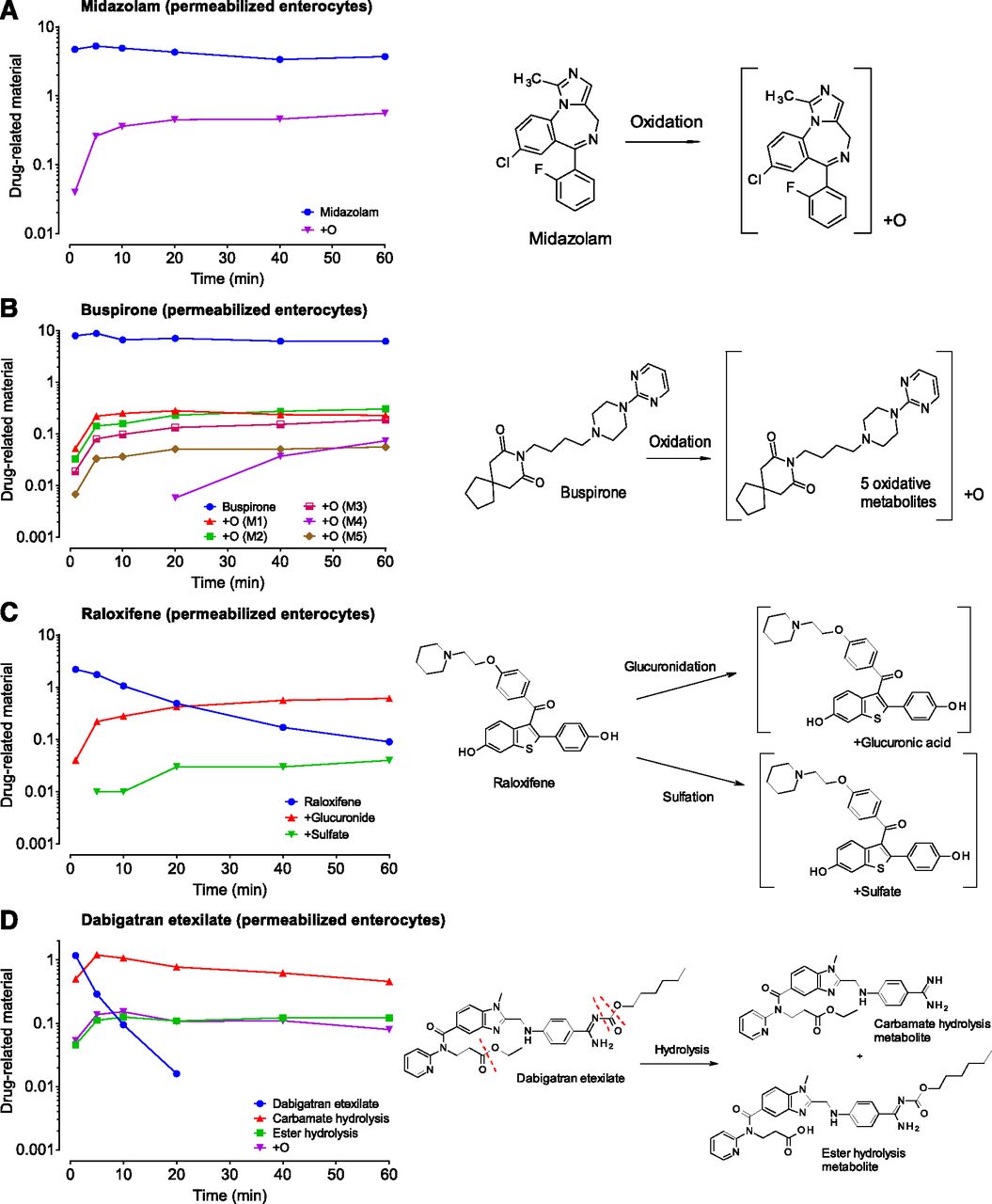
We have analyzed our in vitro samples with QuanQual bioanalytical approach to simultaneously measure both parent compound disappearance and metabolite appearance. In contrast to compound disappearance data used to calculate CLint, since metabolites authentic standards were not used, obtained metabolite formation data are predominantly qualitative. Four compounds were selected to exemplify the utility of this approach: midazolam, buspirone, raloxifene, and dabigatran etexilate. Example data for incubations with permeabilized enterocytes, together with corresponding biotransformation schemes, are presented in Fig. 3 (remaining results for intestinal microsomes and mucosa are presented in Supplemental Fig. 7).

Examples of biotransformation pathway analysis via the QuanQual approach (LC-HRMS). Although midazolam (A) and buspirone (B) predominantly form oxidative metabolites, raloxifene (C) undergoes conjugative metabolism (glucuronidation and sulfation), and dabigatran etexilate (D) is hydrolyzed (at the carbamate and amide functionalities). The y-axis units, drug-related material, represent LC-HRMS peak areas of either parent drug or corresponding drug metabolites. Because authentic standards were not used, presented data should be regarded as mainly qualitative.
Midazolam, a positive control for CYP3A4 metabolism used in all of our assays, showed formation of one prominent +O metabolite, likely corresponding to the 1′-OH-midazolam, the most abundant oxidative metabolite of midazolam at low substrate concentrations (Gorski et al., 1994) (Fig. 3A). Several other metabolites of midazolam were observed at the lower level, including +O (likely a 4-OH midazolam), +O-2H, and +2O metabolites (unpublished data). Buspirone, a dopaminergic anxiolytic and CYP3A4 substrate (Zhu et al., 2005), showed formation of five distinct +O metabolites in human enterocytes, consistent with previous reports (Gammans et al., 1986; Zhu et al., 2005) (Fig. 3B).
Raloxifene, a bis-phenolic estrogen receptor modulator, is known to be both glucoronidated (Kemp et al., 2002; Dalvie et al., 2008) and sulfated (Falany et al., 2006). Both types of metabolites were observed in our incubations with human enterocytes (Fig. 3C), showing that key conjugative biotransformation pathways are present. Raloxifene sulfation, catalyzed by cytosolic sulfotransferases, was observed in human enterocytes and mucosa but was predictably absent in human intestinal microsomes (Supplemental Fig. 7), highlighting the advantages of cellular models. Dabigatran etexilate is a double prodrug of anticoagulant dabigatran (Laizure et al., 2014) containing a cleavable carbamate and ester functionalities. Both hydrolysis products were detected in our intestinal incubations (Fig. 3D), with CES2-catalyzed hydrolysis of carbamate being significantly more prominent (Laizure et al., 2014). Several other oxidative metabolites of dabigatran were also observed (unpublished data).
Fraction Unbound in the Incubation.
The fu,inc values were measured by RED for all test compounds (n = 32) in permeabilized enterocytes and, for a subset of compounds, in intestinal microsomes (n = 15) and mucosa (n = 8) (Table 3). Direct comparison of obtained fu,inc values showed similar nonspecific binding across three models (Fig. 4A; Supplemental Table 1: one-way ANOVA showed insignificant differences among mean values). As an example, the fu,inc values for midazolam were 0.74, 0.74, and 0.71 in permeabilized enterocytes, intestinal microsomes, and mucosa, respectively (Supplemental Table 1). Therefore, the average fu,inc values from permeabilized enterocytes were considered to be representative for in vitro nonspecific binding and used for further predictions of human fg. Measured fu,inc values also showed a statistically significant negative correlation with logD values (Fig. 4B) (Pearson correlation r = –0.64; 95% CI: –0.8120 to –0.3732; P < 0.0001). Although this correlation may be used for predictive purposes, similar to predictions of fu,inc in human hepatocytes (Austin et al., 2005), the nonlinear regression with quadratic equation resulted in relatively poor outcome ( ; r2 = 0.46). Because the relatively short equilibration time of 4 hours was used for all measurements, readers should note that fu,inc values obtained for moderately bound to highly bound compounds may be less accurate (e.g., terfenadine; fu,inc = 0.05) and should be ideally confirmed with longer equilibration time or orthogonal methods (Riccardi et al., 2015; Chen et al., 2019).
; r2 = 0.46). Because the relatively short equilibration time of 4 hours was used for all measurements, readers should note that fu,inc values obtained for moderately bound to highly bound compounds may be less accurate (e.g., terfenadine; fu,inc = 0.05) and should be ideally confirmed with longer equilibration time or orthogonal methods (Riccardi et al., 2015; Chen et al., 2019).
Predicted human fg values for 32 test compounds based on assays in intestinal microsomes, permeabilized enterocytes, and intestinal mucosa are compared with observed in vivo fg values from the literature
See Table 4 for the accuracy and bias of human fg predictions. Values are presented as mean ± S.E.

A comparison of observed fu,inc values for 32 test compounds in human intestinal microsomes, permeabilized enterocytes, and intestinal mucosa (A). A correlation of average fu,inc with lipophilicity (logD at pH 7.4) (B). The fu,inc values were measured at 10 µM of the substrate after 4 hours of equilibrium with the RED device.
Early Predictions of Human fg Values.
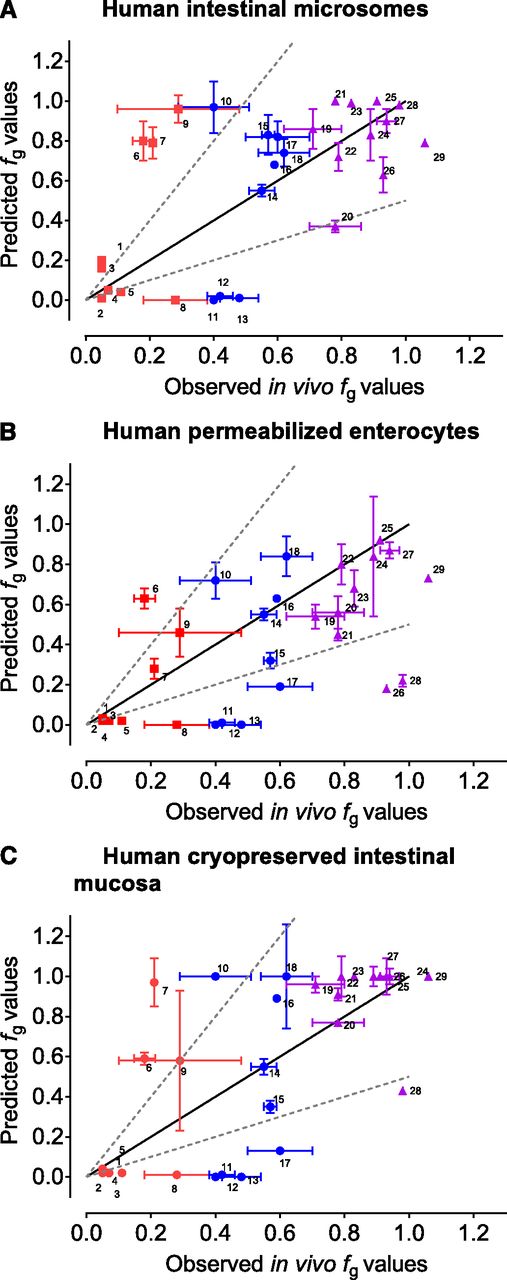
Measured in vitro results (CLint and fu,inc) were supplemented with literature data (passive Caco-2 A→B Papp, Qent, and Aint) and combined using a midazolam-calibrated Qgut model to predict human fg values (see Data Analysis for details). Predicted human fg values were then compared with observed human fg values from the literature (Fig. 5; Table 3). In general, all three in vitro models offered a similar predictivity of human fg values, with GMFE values of 2.62, 2.81, and 2.81 for intestinal microsomes, permeabilized enterocytes, and intestinal mucosa, respectively (Table 4). If compounds are arbitrarily classified according to their observed fg values, the best predictivity was achieved for the high-fg compounds (fg > 0.70; GMFE 1.23, 1.53, and 1.21, respectively). However, the predictivity for low (fg < 0.30; GMFE 4.21, 3.26, and 3.85, respectively) and especially moderate (0.30 < fg < 0.70; GMFE 4.12, 5.06, and 5.76, respectively) fg compounds was significantly inferior (Table 4).

Correlations between observed (in vivo) fg values and predicted fg values based on experiments in human intestinal microsomes (A), permeabilized enterocytes (B), and intestinal mucosa (C). Predicted fg values were calculated based on the midazolam-calibrated Qgut model. Compounds are colored by the range of observed human fg values (0.00–0.30 red ▪▪▪; 0.31–0.69 blue ●; 0.70–1.00 purple ▪▪▪). Values are represented as the mean values ± S.E. Compounds were dabigatran etexilate 1, raloxifene 2, testosterone 3, lovastatin 4, nisoldipine 5, buspirone 6, rifabutin 7, saquinavir 8, simvastatin 9, atorvastatin 10, terfenadine 11, felodipine 12, cyclosporin 13, midazolam 14, cisapride 15, ramipril 16, verapamil 17, sildenafil 18, nifedipine 19, diclofenac 20, methadone 21, zolpidem 22, trazodone 23, repaglinide 24, quinidine 25, indinavir 26, alprazolam 27, benzydamine 28, and enalapril 29.
Accuracy (GMFE and classification success) and bias of human fg predictions based on in vitro assays in three intestinal models
Predictions of human fg can also be evaluated from the perspective of classification success (i.e., low, moderate, and high) and prediction bias (Table 4). Similar to GMFE, classification success analysis showed that only high-fg compounds are acceptably classified based on predictions, especially based on intestinal microsomes (81.8%) and intestinal mucosa (90.9%). The classification success of high-fg compounds based on permeabilized enterocytes was lower (45.5%), with many compounds classified into moderate-fg group instead of high, however. This phenomenon is related to the observed fg underprediction bias in permeabilized enterocytes (i.e., higher metabolism rate) (Table 4). The classification success was acceptable for the low-fg compounds (average 66.7%) but poor for moderate-fg compounds (average 25.9%) (Table 4). Prediction bias was generally not pronounced (Table 4), except in the above mentioned permeabilized enterocytes and high-fg compounds.
The fg predictions for certain compounds varied significantly between different models (e.g., indinavir and rifabutin). Indinavir was significantly underpredicted in permeabilized enterocytes (predicted fg of 0.16 vs. observed fg of 0.91) but better predicted in intestinal microsomes and mucosa (fg of 0.56 and 0.84, respectively). Conversely, rifabutin was well predicted in permeabilized enterocytes (predicted fg of 0.40 vs. observed fg of 0.21) but was poorly predicted in intestinal microsomes and mucosa (fg of 0.79 and 0.93, respectively).
Saturation and Enzyme Kinetics Experiments.
Because all CLint measurements were initially performed at 1 μM of the test substrate, which is typical of drug discovery screening assays, further experiments were performed to evaluate the effects of enzyme saturation on metabolic rates and accuracy of human fg predictions. These experiments were performed with permeabilized enterocytes and two test substrates: indinavir (fg underpredicted by 72%) and raloxifene (fg underpredicted by 2%). Two example compounds were selected based on accuracy of initial fg predictions and diversity of biotransformation pathways (oxidative and conjugative metabolism for indinavir and raloxifene, respectively). In addition to CLint, metabolite formation data were used to estimate the apparent Km values. Results show that indinavir exhibits pronounced saturations of its metabolism (Fig. 6A), with apparent CLint decreasing 10-fold within the concentration range of 0.1–5 μM (from 32.24 to 3.35 μl/min per 106 cells). The apparent CLint at higher concentrations of indinavir (>5 μM) was undetectable (<1 μl/min per 106 cells). The apparent Km value of indinavir oxidative metabolism, based on the sum of all observed metabolites, was 1.24 μM (Fig. 6B). In contrast to indinavir, metabolism of raloxifene showed less saturation, with relatively modest 2-fold decrease of the observed CLint at 0.1–5 μM (from 234 to 125 μl/min per 106 cells; Fig. 6C) and apparent Km value of conjugative metabolism of 8.4 μM (glucuronidation and sulfation) (Fig. 6D). Further increase of raloxifene concentration resulted in additional decrease of observed CLint, with lowest value of 3.24 μl/min per 106 cells at 100 μM of the substrate (Fig. 6C). The Km values determined with an alternative method directly from the substrate disappearance data (Obach and Reed-Hagen, 2002) were comparable to metabolite formation approach [indinavir Km = 0.63 ± 0.11 µM (mean ± S.E.), 95% CI 0.36–1.12 µM; raloxifene 4.73 ± 0.67 µM (mean ± S.E.), 95% CI 3.31–6.69 µM]. Taken together, these results suggest that in-depth evaluations of intestinal drug metabolism should take into account luminal concentrations of the drug rather than nominal concentrations of 1 µM. The challenge is, however, that human luminal drug concentrations are poorly understood at drug discovery stage, at least prior to knowledge of thermodynamic (biorelevant) solubilities and predictions of human efficacious dose. Therefore, assays at 1 µM of substrate are still valuable in discovery settings, especially considering that hepatocyte incubations and/or enzyme phenotyping assays are also often performed at these concentrations.

Enzyme saturation experiments performed in permeabilized enterocytes to assess enzyme saturation as a reason for underpredictions and overpredictions of the human fg. (A and C) show substrate depletion of indinavir and raloxifene at increasing concentrations of the compound. (B and D) show the rate of metabolite formation of indinavir and raloxifene with increasing substrate concentrations (sum of all observed metabolites used). The apparent KM values of indinavir oxidation and raloxifene conjugation were calculated from both substrate disappearance (0.63 and 4.73 µM, respectively) and metabolite formation data (1.24 and 8.43 µM, respectively). Because drug metabolites were not quantified with authentic standards, obtained Vmax values are qualitative and for relative comparisons only.
Discussion
Early predictions of human drug disposition from in vitro data are important in both qualitative and quantitative drug discovery, especially for drug design, safety evaluation, and timely progression of successful molecules. Although intestinal drug metabolism may have a significant impact on drug disposition, it is often studied in either intestinal microsomes (Peters et al., 2016; Hatley et al., 2017a), a model that lacks many drug-metabolizing enzymes, or tissue slices (de Graaf et al., 2010; Li et al., 2016) and organotypic cultures (Almeqdadi et al., 2019) that are rarely available in drug discovery settings (Fig. 1). To address the existing gap, we have compared intestinal microsomes with two emerging models of the human intestine: permeabilized enterocytes and cryopreserved mucosa.
Evaluation with a set of 32 test compounds (Table 1) showed that common CYP- and UGT-mediated metabolism is well captured by all three models (Fig. 2; Table 2). On average, however, enterocytes and mucosa exhibited a 2-fold higher CLint for CYP-mediated reactions, enabling higher sensitivity for the low-clearance compounds. The reasons behind higher CYP-mediated metabolism in enterocytes and mucosa are unclear at this point but may include higher CYP expression in tested batches, better preservation of enzymatic activities, partially different gut regions, or combination of all. Further studies will be needed to address these points, especially prior to broader adoption of these models in drug discovery. In addition, CYP activities compared with intestinal microsomes, enterocytes, and mucosa showed higher rates of CES2-catalyzed dabigatran etexilate hydrolysis (Laizure et al., 2014), FMO-catalyzed benzydamine oxidation, and sulfation of raloxifene and testosterone (Falany et al., 2006), which are indicative of a generally broader application scope. Permeabilized enterocytes may have the additional benefits of enabling assays with poorly permeable or cytotoxic compounds as well as enabling enzyme phenotyping by differential application of cosubstrates (Li et al., 2018b). Metabolic clearance of angiotensin-converting enzyme inhibitors ramipril and enalapril, which are predominantly hydrolyzed in the human liver (MacFadyen et al., 1993; Levitt and Schoemaker, 2006), was low in all tested in vitro intestinal systems. Similarly, AO-mediated oxidation of carbazeran was low in all systems, consistent with reported low expression level of this enzyme in the intestine (Moriwaki et al., 2001). Some of the differences in observed CLint values may be attributed to partially different regions of the human gut from which tested models were prepared (see Material and Methods for details). Although human intestinal microsomes and permeabilized enterocytes were predominantly from duodenum and jejunum, respectively, cryopreserved mucosa was prepared from all three intestinal segments (duodenum, jejunum, and ileum).
In addition to CLint measurements, QuanQual bioanalytical approach enabled direct observation of the metabolite formation and corresponding biotransformation pathways (Fig. 3). Based on examples of midazolam, buspirone, raloxifene, and dabigatran etexilate, this approach can identify key biotransformation pathways, including reactions of oxidation, hydrolysis, and conjugative metabolism. The qualitative profiles also provide important confirmation of the relevant metabolic capacity of these systems. Even if metabolite formation data based on LC-HRMS ionization response are qualitative, or semiquantitative at best (Blanz et al., 2017), this information is invaluable in drug discovery to guide compound design, identify challenges of in vitro–in vivo correlation (e.g., involvement of hydrolytic enzymes or UGTs), or directly compare and contrast intestinal metabolic profiles with liver and other relevant tissues. As an example, early identification of hydrolytic drug metabolism, which is often responsible for (unpredictable) high systemic clearance or prodrug conversion (Nishimuta et al., 2014; Di, 2019), is very important for drug design in lead generation and optimization before significant resources are invested in selected chemotype. Advances in mass spectrometry, especially nanospray techniques (Valaskovic et al., 2006; Schadt et al., 2011) and computational prediction of ionization response (Cramer et al., 2017), may be able to overcome analytical challenges in the future and provide quantitative data on drug metabolites even without authentic standards.
As shown by the equilibrium dialysis experiments (Fig. 4A; Supplemental Table 1), the in vitro nonspecific binding was similar across the three models and significantly correlated with lipophilicity (Fig. 4B). Although such an approach was often historically used to estimate liver fu,inc (Austin et al., 2002; Kilford et al., 2008), the predictivity of intestinal fu,inc from the simple logD correlation was limited, however, and experimental determination is recommended for important compounds. The fraction unbound in the gut is essentially unknown at the drug discovery stage and, similar to previous studies (Gertz et al., 2010), we have assumed a value of 1.
To attempt an early prediction of human fg values, experimentally measured values (CLint and fu,inc) were combined with literature data (Caco-2 A→B Papp, Qent, and Aint) using a midazolam-calibrated Qgut model (Fig. 5; Table 3). Results showed that early predictions of human fg values are feasible, especially for compounds with the high observed human fg values (arbitrarily fg > 0.70; GMFE was 1.23, 1.53, and 1.21 for microsomes, enterocytes, and mucosa, respectively) (Table 4). Because most design teams and discovery projects aim for metabolically stable compounds with high-fg values and good oral bioavailability, this may be a valuable approach to perform early chemotype evaluations. The predictivity of low and moderate human fg values remained a challenge, however, with average GMFEs of 3.77 and 4.98, respectively (Table 4). Although prediction accuracy in the low-to-moderate–fg range is inadequate for decision making, it is sufficient to identify compounds at high risk and/or potential prodrug candidates. Overall, the presented approach may be useful for early assessments of the chemotype, with human fg values >0.7 being well predicted and fg values <0.7 suggesting further evaluation. Based on relatively poor predictivity of low-to-moderate–fg values, authors would recommend against automatic avoidance of flagged compounds, many of which may show acceptable bioavailability after further evaluation. Additional experiments may include enzyme saturation, active transport, biorelevant solubilities, and application of physiologically based pharmacokinetic (PBPK) approaches. These results are generally similar to previous applications of the Qgut model (Yang et al., 2007; Gertz et al., 2010; Karlsson et al., 2013).
Apart from variabilities arising from gut biology (e.g., drug transporters, transit times, and interindividual variability), clinical study design (e.g., number, age, sex, and disease state of subjects), and pharmacological treatment (e.g., drug dose and solubility), the accuracy of fg predictions is also partially biased by the mathematical structure of the Qgut model in relation to CLint (see Materials and Methods for equations). Namely, if CLint is high, the fg is directly proportional to Qgut and inversely proportional to CLint ( . Therefore, the errors of Qgut and CLint will translate into corresponding errors of fg. Conversely, if CLint is low, fg will be close to 1, and errors of CLint and Qgut will play a lesser role (
. Therefore, the errors of Qgut and CLint will translate into corresponding errors of fg. Conversely, if CLint is low, fg will be close to 1, and errors of CLint and Qgut will play a lesser role (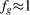 ). This bias of the Qgut model can explain our “apparent” better predictivity at the high observed fg values.
). This bias of the Qgut model can explain our “apparent” better predictivity at the high observed fg values.
To demonstrate the potential impact of enzyme saturation on fg predictions, which are important at higher dose levels of well soluble compounds, example studies with indinavir and raloxifene were performed in permeabilized enterocytes (Fig. 6). Our results confirmed that indinavir metabolism by CYP3A4/5 is highly saturable (Koudriakova et al., 1998), leading to underprediction or overprediction of the intestinal metabolism depending on the concentrations achieved in the intestinal lumen. In addition to demonstrated saturation effects, the predictions of indinavir fg (observed value of 0.93) were model-dependent: 0.63, 0.18, and 0.99 in intestinal microsomes, enterocytes, and mucosa, respectively. The saturation of raloxifene metabolism was less pronounced, however, indicating less uncertainty by performing initial assays at 1 μM of the substrate. The Km values determined based on substrate disappearance and metabolite formation were comparable (within 2-fold), providing both possibilities to drug discovery teams.
Because several efflux and uptake transporters are found in the small intestine (Muller et al., 2017; Oswald, 2019; Couto et al., 2020), drug transport may also impact observed human fg values (Shugarts and Benet, 2009; Yau et al., 2017). For example, our test compounds indinavir and atorvastatin are known substrates for P-gp and breast cancer resistance protein (Estudante et al., 2013), respectively, efflux transporters that may lower the effective permeability and enterocytic intracellular concentrations. Conversely, atorvastatin, enalapril, and verapamil are substrates for organic anion–transporting polypeptides, peptide transporter 1, and organic cation transporters, respectively (Estudante et al., 2013), uptake transporters that may increase effective permeability and facilitate interactions with drug-metabolizing enzymes. In vitro, passive permeability and active transport are not rate-limiting in intestinal microsomes and permeabilized enterocytes, models that lack any significant barrier function, but may play a role in intestinal mucosa because of the presence of intact plasma membrane and transporters yet to be fully evaluated (Li et al., 2018a). Indeed, models like intestinal mucosa may be used in the future to evaluate the interplay between drug metabolism and transport; for example, in the case of dabigatran etexilate, both CES (Bernier et al., 2019) and P-gp (Laizure et al., 2014) play significant roles. This application, however, will require further investigation, both with respect to expression and function of intestinal transporters in these models of human gut.
Considering the relatively low predictivity of the human fg based on the Qgut model (Table 4) independent from the applied in vitro model of the human gut, one may wonder whether static gut models will be useful in the future of drug discovery. The Qgut model is based on simple assumptions: It represents the gut as a single homogenous compartment with uniform permeability, blood flow, and distribution of enzymes while not considering the role of transporters and pharmacological treatment (e.g., drug dose and solubility). The future of drug discovery, therefore, will likely require PBPK models that can account for additional disposition mechanisms, including biorelevant solubilities, enterocytic concentrations, saturation of metabolism, and active transport. Indeed, an early application of intestinal PBPK model improved fg predictions for indinavir, saquinavir, and terfenadine over the Qgut model (Gertz et al., 2011). Similarly, advanced dissolution absorption and metabolism (Darwich et al., 2010) and advanced compartmental and transit models (Gobeau et al., 2016) showed promise in drug discovery applications. The usefulness of intestinal PBPK models in drug discovery will be advanced with refined understanding of the gut physiology, especially with abundance of relevant enzymes and transporters (Couto et al., 2020) and computational approaches (i.e., machine learning/artificial intelligence) to predict compound properties based on the chemical structure alone—for example, biorelevant solubilities (Boobier et al., 2017) and transporter liabilities (Jain and Ecker, 2019).
In conclusion, we have evaluated three models of human intestine from the perspective of drug discovery. All three intestinal models showed reliable CYP and UGT enzymatic activities, but enterocytes and mucosa may offer advantages for low-clearance compounds and alternative biotransformation pathways (e.g., sulfation, hydrolases, and FMO oxidation). A number of knowledge gaps will need to be addressed in the future, including protein expression of drug-metabolizing enzymes and transporters and its overall predictivity of clinical outcomes. For the moment, applications of these models together with the QuanQual bioanalytical approach and midazolam-calibrated Qgut model enabled both qualitative (biotransformation pathways) and quantitative (CLint and early fg estimate) compound evaluation at the early stage, bringing together valuable information for drug design and discovery teams.
Acknowledgments
Authors would like to acknowledge University of Bath and Cogent Skills for support with student placement program. Vishal Ranglani is acknowledged for help with RED assay. Anna-Lena Ungell, Jamie Henshall, Peter Theil, and Emre Isin are greatly acknowledged for their support and encouragement. Lisa Jödicke is acknowledged for help with preparation of illustrations in Fig. 1. Albert Li and Behzad Emadi-Khiav from In Vitro ADMET Laboratories, LLC are acknowledged for help and support with human permeabilized enterocytes and cryopreserved mucosa.
Authorship Contributions
Participated in research design: Davies, Peramuhendige, Golding, Penney, Manevski.
Conducted experiments: Davies, Peramuhendige, Kotian, Shah, Manevski.
Contributed new reagents or analytic tools: Peramuhendige, Kotian, Shah.
Performed data analysis: Davies, Peramuhendige, King, Penney, Shah, Manevski.
Wrote or contributed to the writing of the manuscript: Davies, Peramuhendige, King, Penney, Shah, Manevski.
Footnotes
- Received May 8, 2020.
- Accepted August 7, 2020.
↵1 Current affiliation: F. Hoffmann-La Roche Ltd., Basel, Switzerland.
This work and its authors were funded by UCB CellTech UK, 216 Bath Rd., Slough SL1 3WE, United Kingdom.
Abbreviations
- A→B
- apical to basolateral
- Aint
- intestinal surface area
- AO
- aldehyde oxidase
- CES
- carboxylesterase
- CI
- confidence interval
- CLint
- intrinsic clearance
- fa
- fraction absorbed
- fg
- fraction escaping gut metabolism
- fu,inc
- fraction unbound in the incubation
- FMO
- flavin-containing monooxygenase
- GMFE
- geometric mean fold error
- HRMS
- high-resolution MS
- LC
- liquid chromatography
- MS
- mass spectrometry
- Papp
- apparent permeability
- PBPK
- physiologically based PK
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- Qent
- enterocytic blood flow
- QuanQual
- quantitative-qualitative bioanalysis
- RED
- rapid equilibrium dialysis
- UDPGA
- uridine 5′-diphosphoglucuronic acid trisodium salt
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}