


Abstract
In vitro systems such as cultured hepatocytes are used early in drug development as a proxy for in vivo data to predict metabolites in human and the potential preclinical species. These data support preclinical species selection for toxicity studies as well as provide early evidence for potential active and reactive metabolites that can be generated in human. Although in vivo data would be best to select preclinical species for a given compound, only in vitro systems are available when selecting toxicity study species. However, as with any in vitro system, the correlation to actual in vivo results can be variable. Understanding the reliability of predicting in vivo metabolites from the various available in vitro assays and determining which system may be most predictive would help de-risk drug development teams’ selection process. In this manuscript, we address these questions: can in vitro systems predict circulating metabolites? If so, is predictivity quantitative or indicative of what levels may be seen circulating? Of the currently available in vitro systems, is one better than the others at generating predictive metabolites? To address the first two issues (general in vitro/in vivo predictivity, and whether any in vitro/in vivo correlations are quantitative), we used historical data from Abbott/AbbVie to compare in vitro metabolite profiles with metabolite profiles from in vivo absorption, distribution, metabolism, excretion, and clinical studies. In this retrospective analysis of historic metabolite profiling data, in vitro systems predicted ∼50% of circulating metabolites present in vivo, across preclinical species and human, with no correlation between apparent concentrations in vitro versus in vivo. To address the final question, we selected 10 commercially available compounds with published metabolism data and incubated them in five common in vitro systems (microsomes, liver S9, suspension hepatocytes, HμREL cocultured hepatocytes, and hepatocyte spheroids); the new in vitro metabolite profiling data were compared against published in vivo data to determine whether any individual system was more accurate in generating known major human circulating metabolites. Suspension hepatocytes and cocultured hepatocytes marginally outperformed the other systems. Current in vitro systems have value early in development when in vivo studies are not feasible and are required for regulatory filings to support preclinical toxicology species selection but should not be treated as wholly representative of a given drug’s in vivo metabolism.
SIGNIFICANCE STATEMENT This is a comprehensive assessment of historic metabolism data quantitating the success rate of in vitro to in vivo predictivity. Reliability of in vitro systems for metabolite profiling is important for early drug development, and understanding predictivity will help give appropriate context to the data. New data were also generated to compare common in vitro liver models to determine whether any could be definitively identified as more predictive of human circulating metabolites than others.
Introduction
Preclinical species testing in the pharmaceutical industry is performed to ensure human safety of new medicines. While submitting an investigational new drug application, the applicant is required to establish that the compound is safe for preliminary testing in humans. At the pre-investigational new drug application stage, in vitro cell-based assays facilitate selection of the preclinical species used in toxicology studies based on human metabolite coverage from each species. In vitro metabolism studies are fast, low cost, easy to conduct, and can be used to study drugs that are not yet approved for humans. In vitro models reduce the number of animals used in testing and are employed to predict circulating metabolites to alleviate metabolites in safety testing (MIST) concerns in early stages of drug development (https://www.fda.gov/media/72279/download). Subsequent in vivo studies performed while the drug candidate is in phase I and phase II will highlight differences in the circulating metabolites in the preclinical species and humans. Human metabolites that can raise safety concerns are those present at greater than 10% of total drug-related exposure at steady state (https://www.fda.gov/media/72279/download). The in vivo studies will either support the preclinical species selected or indicate that another species may be required to demonstrate human safety. If an animal species cannot be identified, then the metabolite requires further safety evaluation requiring additional time and investment for the applicant (https://www.fda.gov/media/72279/download).
In vitro systems, such as cultured hepatocytes, are used early in drug development as a proxy for in vivo data to predict metabolites in human and potential preclinical species (Wang et al., 2010; Burton et al., 2018). However, as with any in vitro system, the correlation to actual in vivo results can be variable. Understanding the metabolic in vitro predictivity of a given assay for in vivo metabolism would allow drug development teams to weigh the significance of early cross-species metabolite profiles before the eventual clinical outcomes. Although it can be beneficial to predict metabolism in all biological matrices (e.g., blood, plasma, bile, urine, tissues), those found to be circulating are of utmost importance for establishing human safety (Iwatsubo et al., 1997; Anderson et al., 2009; Dalvie et al., 2009; Loi et al., 2013; Iegre et al., 2016).
Although the liver is the primary location for xenobiotic metabolism, it is not the only tissue that contains drug-metabolizing enzymes; thus, hepatocyte cultures do not account for all potential metabolic pathways (Krishna and Klotz, 1994; Kapitulnik and Strobel, 1999). Cultured cells also do not perfectly mimic the activity of enzymes in situ, and metabolic profiles generated from intact tissues can differ in both composition and intensity. There is also no guarantee that any given metabolite, even if generated in vivo and/or in hepatocyte cultures, will be circulating in the blood, and thus, of interest to regulatory agencies for MIST consideration.
To assess how effectively in vivo metabolites are predicted by in vitro systems, a review was performed on AbbVie/Abbott historical data consisting of in vitro (combination of hepatocytes, liver microsomes, liver S9, and liver slices) and in vivo metabolite profiles (plasma, bile, and urine when possible). The in vitro systems employed were dependent on the preferred matrix and detection method at the time the study was conducted. Because available historic data includes a variety of in vitro methods as available systems change and improve, we also selected ten commercially available compounds with published human absorption, distribution, metabolism, and excretion (ADME) metabolite data to incubate in various in vitro systems, including liver microsomes, liver S9, suspended hepatocytes, HμREL cocultured hepatocytes, and hepatocyte spheroids. The data were employed for a head-to-head comparison with determine if any of these systems is demonstrably better at predicting circulating metabolites. In addition, data were interrogated to determine if certain biotransformations were more predictive than others and if in vitro results were more representative of any specific biological matrix.
Materials and Methods
Collation of Historic Data
Metabolite identification and biotransformation data from historical studies were used, including metabolite profiling in in vitro assays, in vivo preclinical studies, and clinical samples. For each study, metabolite identities were extracted and linked with metadata including metabolite identity, biotransformation pathway(s), and percent drug-related material (%DRM) represented from radio-data, when available, or mass spectrometry signal response.
Data Filtering and Background Cut-Offs
For radiolabeled studies (in vitro or in vivo), a %DRM cutoff of 1% was set, and any metabolites identified as under 1% DRM were removed from the analysis. Within species, in vitro and in vivo studies were compared for each test article. Metabolites only present in the in vitro system were marked “in vitro only,” metabolites only present in in vivo plasma were marked “in vivo only,” and metabolites detected in both the in vitro system and in vivo were marked “shared.”
Predictivity Calculations
Two different measures of predictivity were calculated: percent shared metabolites relative to in vivo matrix and percent of metabolites unique to the in vitro system.
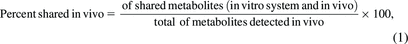 and
and

Incubation of Commercial Test Compounds
Liver Microsome Incubations
The experiments were performed in 96-well plates using pooled liver microsomes. One vial of pooled liver microsomes for each of five species (BioIVT: male CD-1 mouse lot 1313002, male Sprague-Dawley rat lot 1004, male Beagle dog lot 53790, male Cynomolgus monkey lot 53794, and mixed-gender human lot 1710084) was thawed at room temperature.
For the incubation without uridine diphosphate glucuronic acid (UDPGA), the 200 μl incubation mixture contained 1 mg/ml liver microsomal protein in 50 mM phosphate buffer (pH 7.4) and drug compound (final concentration 10 μM). The reactions were started by the addition of NADPH (final concentration 1 mM) and incubated at 37°C in a water bath for 60 minutes. At the end of the incubation period, the reaction was quenched by the addition of 200 μl 1:1 (v/v) acetonitrile:methanol containing 1.5 μM carbutamide as an internal standard. After quenching, all samples were vortexed and centrifuged. An aliquot of 10 μl of each supernatant was injected and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and the rest of the sample was stored at –20°C. For the 0-hour time point, quench solution was added prior to the addition of cofactor.
For the incubation with UDPGA, the 200 μl incubation mixture contained 1 mg/ml liver microsomal protein in 50 mM phosphate buffer (pH 7.4) and drug compound (final concentration 10 μM). The reactions were started by the addition of NADPH (final concentration 1 mM) and UDPGA (final concentration 5 mM) and incubated at 37°C in a water bath for 60 minutes. At the end of the incubation period, the reaction was quenched by the addition of 200 μl 1:1 (v/v) acetonitrile:methanol containing 1.5 μM carbutamide as an internal standard. After quenching, all samples were vortexed and centrifuged. An aliquot of 10 μl of each supernatant was injected and analyzed by LC-MS/MS and the rest of the sample was stored at –20°C. For the 0-hour time point, quench solution was added prior to the addition of cofactors.
Liver S9 Incubations
One vial of pooled liver S9 for each of five species (BioIVT: male CD-1 mouse lot PQT, male Sprague-Dawley rat lot WAE, male Beagle dog lot MNL, male Cynomolgus monkey lot ADO, and mixed-gender human lot VKC) was thawed at room temperature. The pooled liver S9 was first incubated with alamethicin (100 ug/ml) on ice for 15 minutes to allow for pore formation, and then mixed with 50 mM phosphate buffer (pH 7.4), MgCl2 (final concentration 3.3 mM) and compound (final concentration 10 μM). The final volume of the incubation mixture was 200 μl containing 2 mg/ml liver S9. The reactions were started by the addition of NADPH (final concentration 1 mM), glutathione (final concentration 5 mM), and UDPGA (final concentration 5 mM) and incubated at 37°C in a water bath for 60 minutes. At the end of the incubation period, the reaction was quenched by the addition of 200 μl 1:1 (v/v) acetonitrile:methanol containing 1.5 μM carbutamide as internal standard. After quenching, all samples were vortexed and centrifuged. An aliquot of 10 μl of each supernatant was injected and analyzed by LC-MS/MS or stored at –20°C. For the 0-hour time point, quench solution was added prior to the addition of compound.
Suspended Hepatocyte Incubations
One vial of cryopreserved hepatocytes for each of five species (BioIVT: male CD-1 mouse (lot SCQ), male Sprague-Dawley rat (lot WHU), male Beagle dog (lot UHC), male Cynomolgus monkey (lot LSS), and mixed-gender human (lot KDH) was placed for 1.75 minutes in a 37°C water bath until the ice just melted. The contents of the vials were transferred to separate 50-ml conical tubes containing 46.5 ml of prewarmed (37°C) InVitroGRO HT thawing medium. The conical tubes were capped and inverted gently several times to fully thaw the hepatocytes. The tubes were centrifuged (Eppendorf model #5810R) at 100 × g for 5 minutes at room temperature to gently pellet the hepatocytes. Supernatant was poured off and the pellet was gently resuspended and diluted to 1,000,000 viable cells/ml in hepatocyte incubation media (Gibco William’s E Media containing 1× GlutaMax L-glutamine and 15 mM HEPES) at 37°C. Diluted hepatocyte suspension (250 μl) was added to experimental wells (250,000 viable cells/well) of the 24-well plate and into the 0 hours of the deep well quench plate. Blank media (250 μl) was added to the no-tissue control wells of the 24-well plate and quench plate. One milliliter of 1:1 (v/v) acetonitrile/methanol quench solution was added to the 0-hour time points of the deep well plate. Ten microliters of 10 mM stock compound in DMSO was diluted with 5 ml of incubation media and mixed well. The working stock solution (250 μl) was pipetted into appropriate wells of the incubation plate for the 4-hour incubations and to the 0-hour wells of the deep well plate. The quench plate was sealed and stored at 4°C. The 24-well plates were incubated in a Forma Scientific Incubator #3130 at 37°C in 5% carbon dioxide, with gentle shaking (VWR plate shaker) for 4 hours. After 4 hours, the reactions were quenched with the addition of 1 ml 1:1 (v/v) acetonitrile/methanol. The entire well contents from the 4-hour incubation plate were transferred to appropriate wells of the 96-deep well plate. Plates were centrifuged for 30 minutes at 3300 RPM (2179 × g) using an Eppendorf A-4-62 rotor (Eppendorf Centrifuge, model 5810R). The supernatant was removed and transferred to a 96-well assay plate and analyzed by LC-MS/MS.
HμREL Cocultured Hepatocyte Incubations
HμREL hepatic cells from male CD-1 mouse (HU7002MS), male Sprague-Dawley rat (HU3052RT), male beagle dog (HU2012DG), male cynomolgus monkey (HU4009PR), and mixed-gender pooled human (HU1064HUP), cocultured with mouse fibroblast nonparenchymal stromal cells, were purchased from HμREL Corporation (Newark, NJ). On receipt of HμREL plates, maintenance media was warmed to 37°C in a treated water bath. The lid was removed, and sealing film was peeled off using aseptic technique. Using low vacuum pressure, shipping media was gently aspirated making sure to not contact the bottom of the wells. The wells were replenished with serum-containing maintenance media at 500 μl/well. A sterile replacement lid was placed on the 24-well plate and the plate was transferred to a sanitized incubator (Forma Scientific) set at 37°C and 5% CO2 with a treated water pan located at the bottom shelf to maintain humidity. HμREL hepatic coculture plates were incubated overnight.
After 24 hours (day 0), HμREL dosing media was warmed to 37°C, and the maintenance media was aspirated in the same manner as the shipping media. Compounds were diluted with dosing media in sterile reservoirs to obtain a final concentration of 10 μM of compound. Diluted compound in dosing media (500 μl) was transferred to corresponding wells for all five species (mouse, rat, dog, monkey, and human) plus a stromal cell line for 3- and 7-day time points. Timolol (A-67167) was incubated for 3 and 7 days as a positive control. After the incubations were complete, the appropriate wells were quenched with 1 ml methanol/acetonitrile (1:1 v/v) containing 1.5 μM carbutamide. The entire contents of the wells were transferred to a 96-deep well plate and centrifuged for 30 minutes at 2192 × g (A-4-62 rotor and model 5810R centrifuge, Eppendorf) prior to analysis.
Hepatocyte Spheroid Incubations
Cryopreserved primary human hepatocytes (BioIVT, lot #ZSE, lot #CRT) were used for forming spheroid cultures. Cells were thawed in spheroid spin media (BioIVT INVITROGRO Spheroid Spin Medium, cat. no. Z990032) at 37°C and were transferred into warmed spheroid plating medium (BioIVT, cat. no. Z990033) per the manufacturer’s protocol. Following centrifugation at 100 × g, cells were resuspended in spheroid plating medium to a final concentration of 632,000 cells/ml. Cells were seeded at 31,600 viable hepatocytes/well into Corning Elplasia 96-well round bottom ultra-low attachment microcavity microplates (cat. no. 4442) per the manufacturer’s protocol and were subsequently centrifuged at 100 × g for 2 minutes to yield 400 viable cells per spheroid. Spontaneous self-aggregation of the hepatocytes produced spheroid formation over 5 days of culture (37°C, 5% CO2) with a 50% media change every 48 hours. After 5 days post seeding, 50% of the media was removed from each well and was replaced with serum-free spheroid maintenance medium (BioIVT INVITROGRO, cat. no. Z990034) three times. On the final rinse, test compounds diluted to 20 μM in spheroid maintenance medium were added to their respective test wells to a final concentration of 10 μM. Plates were incubated (37°C, 5% CO2) for 0, 3, or 7 days in the absence of media changes. At the end of each time point, spheroid cultures were quenched with 1:1 acetonitrile:methanol containing 50 nM carbutamide and samples were prepared for LC-MS/MS analysis. For the 0-day time point, quench solution was added prior to the addition of compound. Compound-only incubations in the absence of hepatocyte spheroids served as controls for metabolism.
Results
Overview of Dataset and General Metrics
The retrospective analysis investigated the predictive capability of in vitro assays (typically suspension hepatocytes, but also some microsomes and cocultured hepatocytes) (Table 1) for in vivo results (from radiolabeled ADME studies, first-in-human studies, and preclinical toxicology studies). The various assays representing the in vitro data were grouped into a single data set. When available, radioactivity data were used to quantitate metabolites and normalize the percent drug-related material (%DRM) in each sample for comparison across biological matrices and analytical platforms. All other studies used mass spectral response for quantitating each metabolite and calculating %DRM (note that due to potential difference in ionization efficiency between parent and metabolites, quantifying in this way may be less accurate than using radioactivity signal). A total of 138 studies were investigated, encompassing 18 human, 10 mouse, 90 rat, 16 dog, and 4 monkey studies (Table 1). Mass spectrometry data were primarily obtained from Thermo Orbitrap mass spectrometers, including LTQ-Orbitrap, QExactive, and Fusion Tribrids. A summary of the average number of metabolites detected in circulating profiles versus in vitro profiles is available in Table 1; in general, more metabolites are produced in vitro than are present circulating, but there is significant variability in metabolite count across the whole of the data.
A summary of the historic data used for the retrospective analysis
Dataset is broken down by species, in vitro system, and number of metabolites identified. Columns two through four represent the number of in vitro/in vivo study pairs that have liver microsomes, suspension hepatocytes, and cocultured hepatocytes as their in vitro matrix, respectively. Columns six and seven contain the average number of metabolites identified in the in vivo circulating profiles and the in vitro profiles, respectively.
Predictivity Calculations
Understanding the ability of in vitro assays to predict the in vivo results is important for making project related decisions. In this analysis, two calculations facilitate the understanding of the in vitro assays’ predictability for in vivo systems using the historical data. The percentage shared metabolites in plasma represents the proficiency of the in vitro assay to generate the metabolites seen in vivo, true positives (eq. 1).
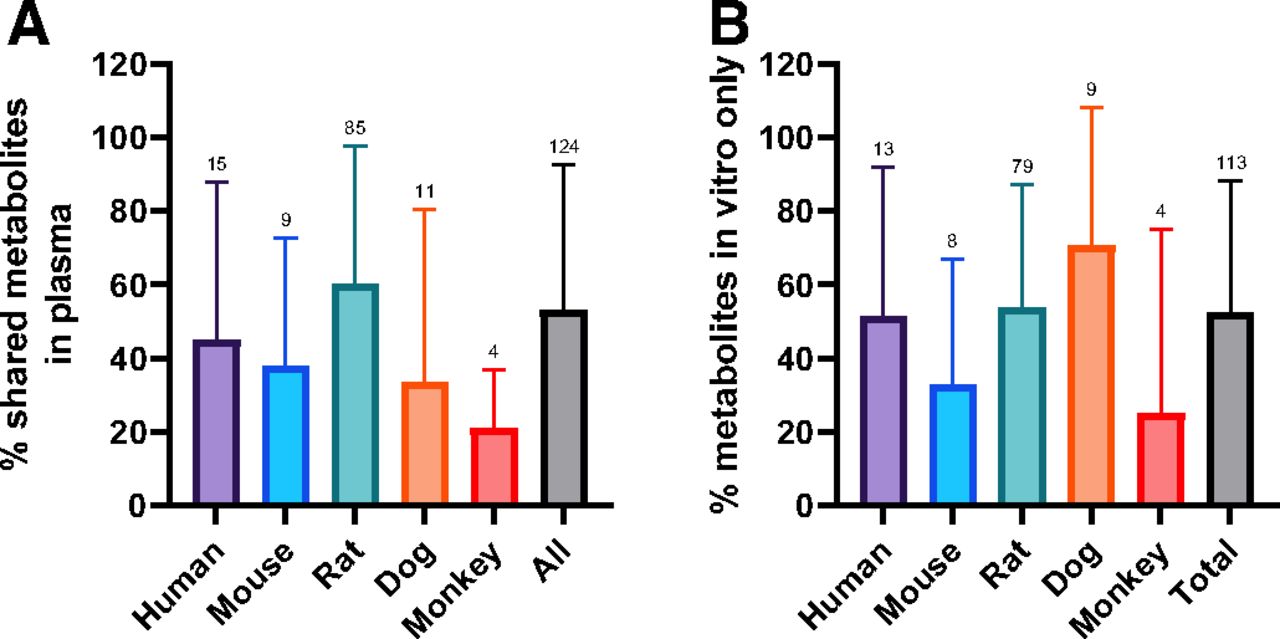
This would be 100% if all the metabolites in vivo were observed in the in vitro assay. An average of 53% of circulating metabolites were covered by in vitro metabolite profiles across all species (Fig. 1A), with a spread from 0% to 100% coverage. The data indicates no major difference between species with human, mouse, rat, dog, having 45%, 38%, 60%, and 33% coverage, respectively, with a significant overlap in standard deviations. Although monkey was lower (average of 21% coverage), the data comprised only four studies. It is also critical to understand the false-positive rate, or the number of metabolites generated by in vitro assays that are not observed in vivo (eq. 2).

Percent of circulating profiles that were successfully predicted by in vitro systems using eq. 1 (A) and percent of in vitro metabolites not detected in vivo in plasma using eq. 2 (B). Error bars represent S.D. An average of 53% of circulating metabolites was predicted by in vitro metabolite profiles, with no major species differences. An average of 53% of metabolites detected in vitro was not detected in plasma. Annotations are the number of studies per analysis. Error bars represent S.D.
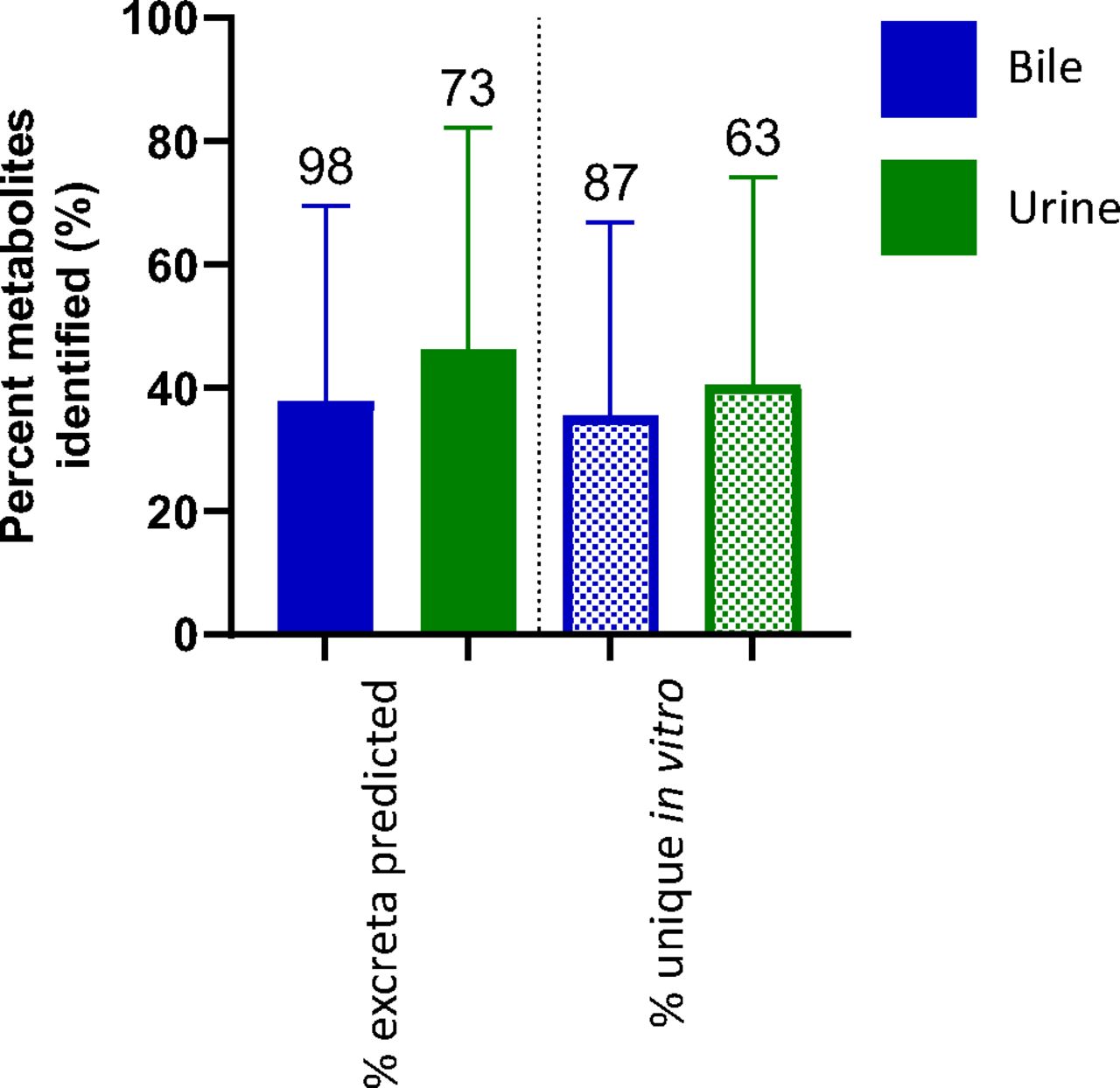
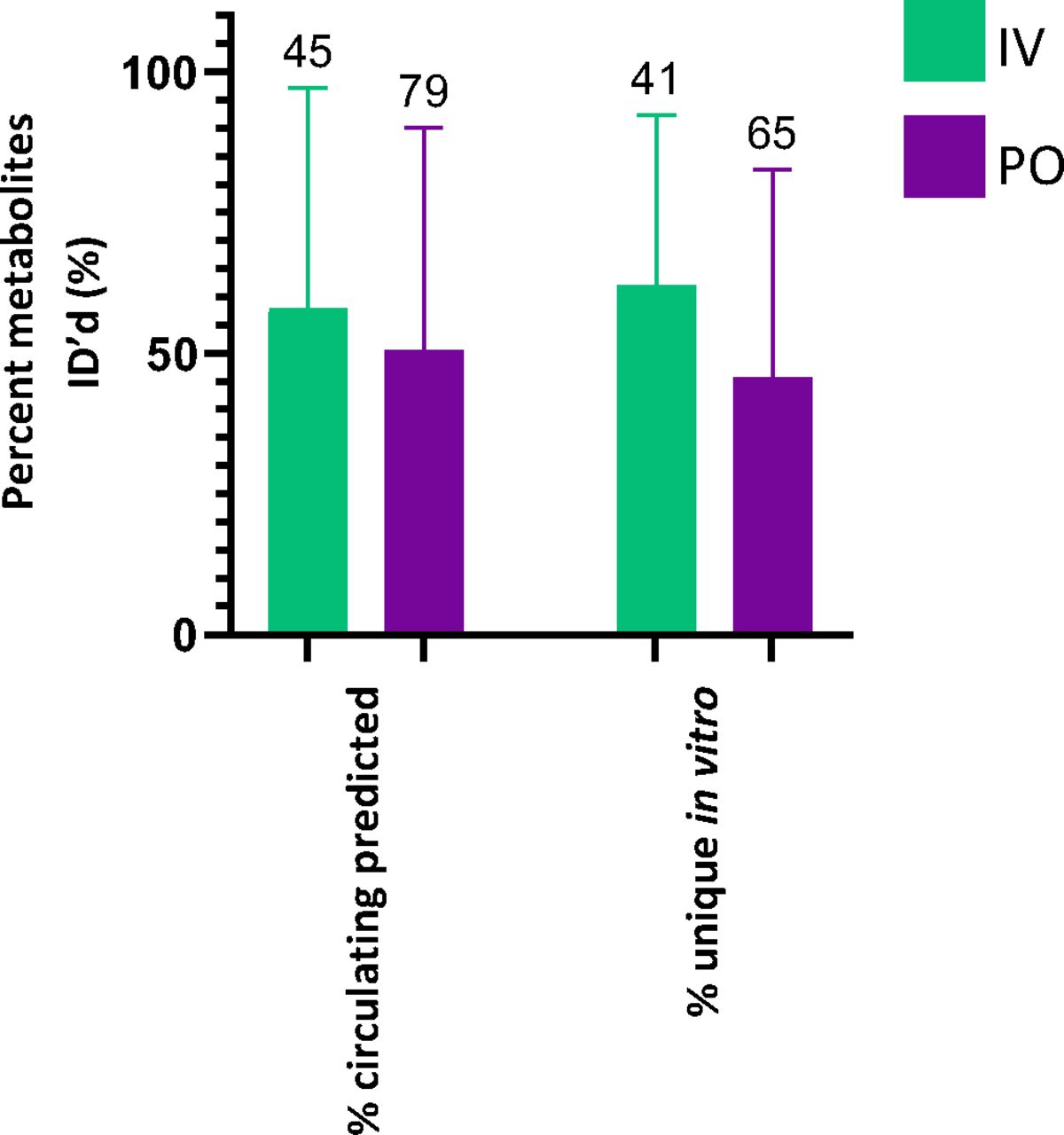
An average of 53% of the metabolites detected in vitro were not detected in circulating profiles (Fig. 1b), with no significant difference between species. The metabolites generated from the rat in vitro assays were also compared with the available excretion data, with an expectation for a higher correlation. Excreta from bile-duct cannulated rat ADME studies did not outperform circulating predictivity. Coverage of rat bile and urine profiles by rat in vitro systems is 38% and 46%, respectively; false-positive rates are similar, averaging 36% in bile and 41% in urine (Fig. 2). A breakdown of the route of administration (oral vs. intravenous), inclusive of all species in the dataset, also exhibited no differences for circulating metabolite predictability (Fig. 3).


Quantitative Predictivity
A common practice in several pharmaceutical companies is assuming the abundant metabolites identified from in vitro assays will be major circulating metabolites, and thus they will initiate metabolite synthesis at an early stage. Do statistics agree with this premise as a good use of resources? The correlations, thus far, categorized successful in vitro/in vivo predictions as the circulating metabolite being present in the in vitro assay, irrespective of peak area. For the studies in which quantitative data (primarily radiolabeled chromatograms) are available, the analyses were taken a step further and metabolite abundance was correlated between in vitro and in vivo assays. Metabolites identified in the in vitro assays were binned into three categories based on individual %DRM (high abundance [>10% DRM], medium abundance [5 <10% DRM], and low abundance [1%–5% DRM]) and compared with the corresponding circulating profile estimations designated as the same high, medium, or low abundance categories. Peaks below 1% DRM in the in vitro studies were not included in the comparison. Of the high abundance in vitro metabolites, 56% were high abundance in vivo whereas 26% and 18% were medium and low abundance respectively. The medium abundance in vitro peaks were estimated to be 65%, 21%, and 14% high, medium, and low abundance, respectively, in vivo. Finally, the low abundance in vitro metabolites were calculated to have 45% high, 26% medium, and 29% low abundance in vivo. Overall, the data indicate poor correlation of in vitro abundance with in vitro abundance. Therefore, it is suggested to wait for in vivo data before synthesizing projected major circulating metabolites rather than spending those resources based on in vitro data that can be misleading and unnecessary.
Predictivity Comparison across In Vitro Systems
Novel in vitro systems have been introduced since the retrospective data were collected. The selection of preclinical species and early understanding of MIST issues could benefit tremendously from the ability to use in vitro assays to predict major metabolites (>10% DRM). Are any of these novel in vitro systems more robust for predicting in vivo metabolites? To test some of these systems, 10 commercially available compounds with known human metabolism (venetoclax, ombitasvir, dasabuvir, ritonavir, alpelisib, linezolid, siponimod, momelotinib, pevonedistat, and clozapine) were selected to investigate how well the various in vitro platforms (liver microsomes, liver S9, suspended hepatocytes, 3D hepatocyte spheroids, and HμREL cocultured hepatocytes) predict major circulating metabolites. These compounds were known to have 15 major metabolites (>10% DRM): eight oxidations, five hydrolysis products, one dealkylation, and one glucuronidation (Dain et al., 1997; Denissen et al., 1997; Slatter et al., 2001; Liu et al., 2016; Shen et al., 2016a, 2016b; James et al., 2017; Glaenzel et al., 2018; Zheng et al., 2018; Bolleddula et al., 2022). The data indicate that no individual system generates all 15 major metabolites across the 10 compounds. Five of the major metabolites were detected in liver microsomal incubations, six were detected in the hepatocyte spheroids, eight in liver S9, eight in suspended hepatocytes, and 10 were covered in the HμREL cocultured hepatocyte incubations (Table 2). Four major metabolites were not detected in any in vitro system; three of which were at least third generation (three separate biotransformation events), including multiple different biotransformation pathways. No major in vivo metabolites of interest were uniquely generated in vitro by liver microsomes, liver S9, or suspended hepatocytes; one major metabolite each was uniquely generated by hepatocyte spheroids (oxidation) and HμREL cocultured hepatocytes (amide hydrolysis). Both unique metabolites were second-generation, requiring two sequential biotransformation events. Based on the true-positive rate (67%), the data indicated that cocultured hepatocytes were the optimal system tested. Hepatocyte spheroids were the next-best at 60% based on the full dataset, but if only metabolites detected in both donors are considered, the success rate drops to 40%, below both suspended hepatocytes and liver S9. Suspended hepatocyte and liver S9 had equivalent success rates, 53%, but hepatocytes covered a broader range of biotransformation pathways, including a second-generation glucuronidation, whereas liver S9 generated only phase I metabolites (oxidation pathways).
A direct comparison of liver microsomes, liver S9, suspended hepatocytes, and two advanced hepatocyte systems—HμREL coculture and spheroids—on the same set of 10 commercially available compounds comprising 15 major circulating metabolites
Trends Relative to Biotransformation Pathways
The data were assessed to identify specific in vivo biotransformation’s that are more predictive or less predictive using the in vitro data. Across all species in the retrospective analysis, 91 in vivo metabolites were described above the inclusion criteria of 1% of dose-related material (%DRM). Of those, 52 were first-generation metabolites (requiring a single biotransformation), and 39 were second-generation or higher (requiring more than one sequential biotransformation); 73 were oxidative or phase I, and 21 were phase II conjugation (e.g., glucuronidation or glutathione conjugation). No marked differences in predictivity were found between phase I (oxidative) and phase II (conjugation reaction) biotransformation pathways—42% of phase I and 48% of phase II circulating metabolites were observed in vitro. However, the in vitro data are more likely to contain first-generation metabolites than metabolites that require more than one biotransformation step (second-generation+): 48% of the first-generation metabolites were detected in the in vitro profiles whereas only 33% of the second-generation or greater metabolites were observed in vitro (Table 3).
Comparison of biotransformation pathways represented across all the historical data sets. Individual metabolites, irrespective of species, were binned by phase (phase I oxidative/reductive metabolism or phase II conjugation) or by generation (number of biotransformation steps)
Metabolites detected in multiple species were only counted once.
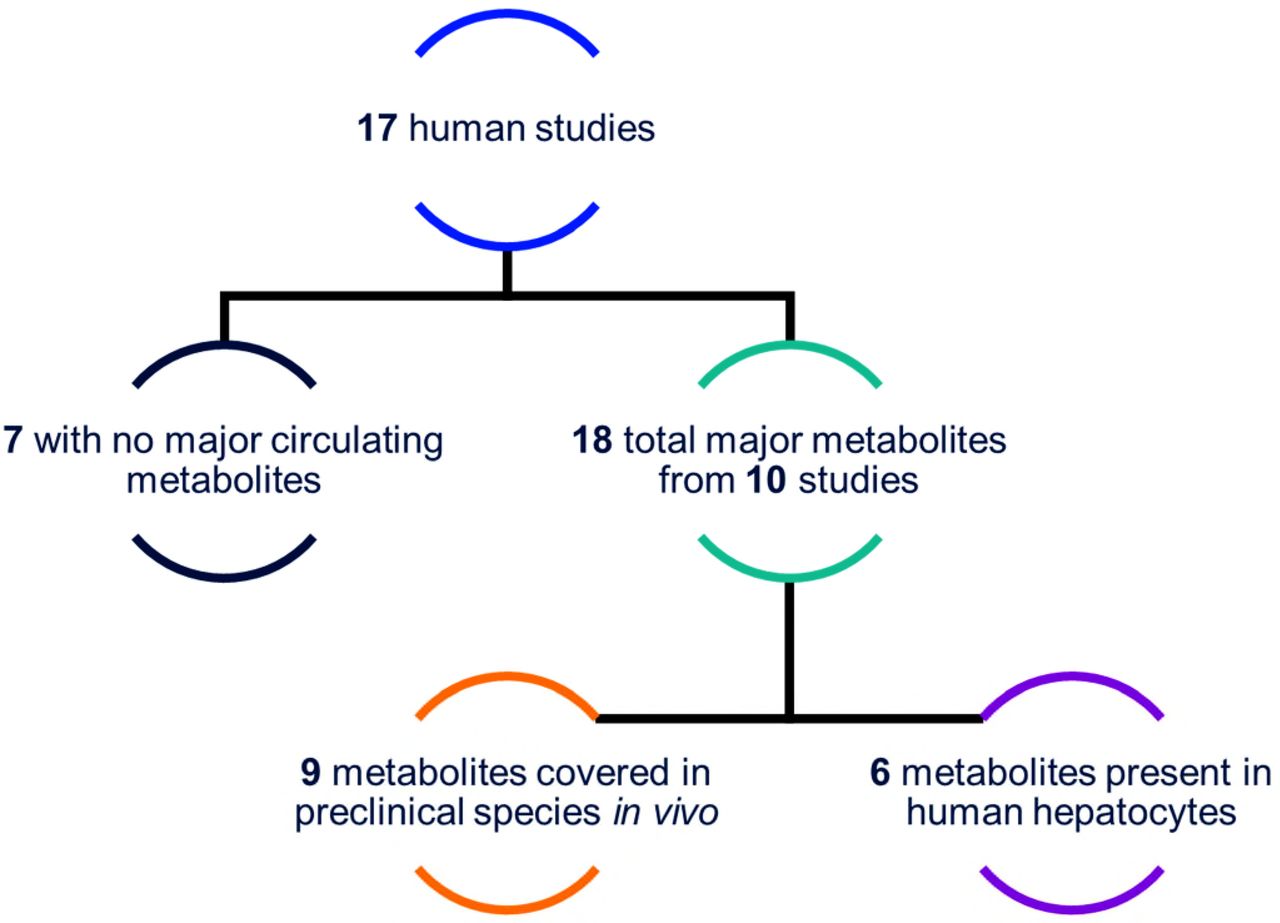
In a specific analysis of human data, summarized in Fig. 4, the historic dataset includes 17 in vivo human metabolite profiling studies that comprise 18 major circulating metabolites (detected above 10% DRM) across 10 different compounds (seven compounds formed no major circulating metabolites). Of those 18 major metabolites, six were predicted by in vitro systems and nine were identified as circulating in preclinical species (not necessarily major in preclinical species). Five of the six in vitro metabolites were single generation metabolites (the sixth was a +O +glucuronide). For those circulating human metabolites not observed in the in vitro systems, 58% were second-generation or greater and 42% were single generation or primary metabolites. In particular, the data demonstrated that in vitro systems did not generate any major amide hydrolysis metabolites. Of the seven compounds that produced no major circulating metabolites, three also resulted in no human in vitro metabolites, whereas the remaining four compounds produced an average of 4.5 human in vitro metabolites.

Graphic summary of in vitro versus in vivo metabolite profiles for 17 compounds in the historic data with available human in vivo metabolite profiles.
Comparing human circulating metabolite profiles revealed that no specific biotransformation pathways were more or less likely to be captured by preclinical species. The metabolites covered in preclinical plasma are roughly equally divided between primary and secondary metabolites, but none of the metabolites of a generation greater than two was predicted.
Discussion
A major goal of in vitro metabolite profiling and in vivo work in preclinical species is to predict and account for human circulating metabolites. Potential for active metabolites must be accounted for in dose projections, and coverage of human circulating metabolites is required by species selected for toxicological assessments—metabolites unique to human are typically concerning, as they require additional study to account for potential active or toxic effects. According to our analysis, the success rate of predicting in vivo circulating metabolites using in vitro metabolite profiling is slightly over 50% for the data described here, irrespective of species or dose route, with a slightly higher success rate in predicting single-step metabolites than those requiring multiple biotransformation steps. Similar assessments from Anderson et al. (2009) and Iegre et al. (2016) found comparable success rates, albeit using smaller test sets. Predictivity of in vitro liver systems for bile and urine metabolites was not markedly better than for circulating metabolites; the false-positive rate of in vitro systems in predicting circulating metabolites is not explained by generation of excreted metabolites, but likely by the generation of metabolites with no in vivo relevance.
In a direct comparison of liver microsomes (+UDPGA), liver S9, suspended hepatocytes, and two advanced hepatocyte systems—HμREL coculture and spheroids—on the same set of compounds, no in vitro system covered all major metabolites for the commercial compounds tests, although HμREL cocultured hepatocytes were the most successful, generating two-thirds of the metabolites of interest. However, it also generated the greatest number of metabolites total, even over hepatocyte spheroids, which can be incubated for the same length of time. More metabolites in vitro may improve the ultimate success in generating major circulating metabolites, but they complicate the process of determining the value of individual metabolites at early stages before in vivo work can be conducted. We did not find any correlation between intensity or %DRM in the in vitro incubations and in vivo abundance, so parsing the true in vivo metabolites from the “extras” in an in vitro experiment is difficult using just the profiling data. Current in vitro systems are not a perfect model for in vivo metabolism but still have value early in development when in vivo studies are not feasible and are required for regulatory filings to support preclinical toxicology species selection. Further study of potential physicochemical properties of the parent drug and its metabolites or other in vitro systems may add value orthogonal to the metabolite profiles alone that could improve in vitro/in vivo translation.
Authorship Contributions
Participated in research design: Freiberger, Thompson, Jenkins, Wagner.
Conducted experiments: Thompson, Zhang, Underwood, Lynch.
Performed data analysis: Freiberger, Thompson, Zhang, Underwood.
Wrote or contributed to the writing of the manuscript: Freiberger, Thompson, Zhang, Underwood, Lynch, Jenkins, Wagner.
Footnotes
- Received May 7, 2024.
- Accepted October 10, 2024.
All authors are employees of AbbVie and may own AbbVie stock. AbbVie sponsored and funded the study; contributed to the design; participated in the collection, analysis, and interpretation of data, and in writing, reviewing, and approval of the final publication.
Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- DRM
- drug-related material
- MIST
- metabolites in safety testing
- UDPGA
- uridine diphosphate glucuronic acid
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}